
«Предком» AlphaFold является алгоритм AlphaGo, который стал играть в го лучше любого человека. Источник: DeepMind
Разработчики из Deep Mind за последние пару лет стали известны благодаря многим своим проектам. В частности, они научили искусственный интеллект (слабую его форму) играть в Go, классические Atari-тайтлы и некоторые другие игры, сложные для «понимания» машиной. Сейчас наступил черед более серьезных занятий — Deep Mind постепенно меняет специализацию ИИ на молекулярную биологию.
Если точнее, то искусственный интеллект учат прогнозировать структуру белка на основе фрагмента последовательности аминокислот — этих кирпичиков белковой жизни. Проект, о котором идет речь, получил название AlphaFold. ИИ научили работать быстрее и точнее, чем люди благодаря обучению по базе последовательностей, собранных генетиками за несколько лет.
В конкурсе Critical Assessment of Structure Prediction (CASP), где необходимо было прогнозировать структуру белка, искусственный интеллект от Deep Mind занял первое место, став лидером среди 98 участников. ИИ удалось правильно предсказать структуру 25 из 43 белков. На втором месте — команда, которой удалось правильно предсказать структуру 3 из 43 белков. В ходе «соревнования» каждой команде ежемесячно отправляли определенный набор аминокислот. Это происходило в течение нескольких месяцев. Команды, получив все элементы, должны были предсказать структуру белка, который составляют эти аминокислоты. Структура ранее была уже определена учеными, так что правильный ответ был у организаторов.
Для науки исследования такого рода имеют первостепенное значение, поскольку белок — это основа жизни. Соответственно, предсказывая структуру белка, можно научиться понимать многие биологические функции и процессы. Стоит отметить, что в некоторых случаях ученые тратят годы на то, чтобы предсказать структуру определенного белка. Проблема в том, что в ДНК обычно есть данные о последовательностях аминокислот, но не структурах, которые формируют цепочки из них.
В организме человека содержится огромное количество разновидностей белка. По разным оценкам, оно может достигать нескольких миллиардов. Структур белка и того больше — количество описывает число с 300 нулями. 3D форма белка зависит от многих факторов — количества аминокислот, длины цепочки и т.п. Пространственная структура определяется еще и ролью, которую выполняет определенный белок в организме человека.
К примеру, клетки сердца строятся при помощи белка, сложенного таким образом, чтобы молекулы адреналина, идущие по кровеносной системе человека, задерживались и ускоряли темп биения сердечной мышцы. Практически любая из способностей и возможностей организма зависит от формы определенного белка — от сокращения мышц до зрения.
Чем сложнее белковая структура, тем сложнее ее моделировать. Стоит отметить, что некоторые болезни, считающиеся проблемой нового века, вызываются ошибочным сворачиванием белковых структур. К таким заболеваниям относятся, в частности, болезнь Альцгеймера, Паркинсона, кистозный фиброз и болезнь Гентингтона.
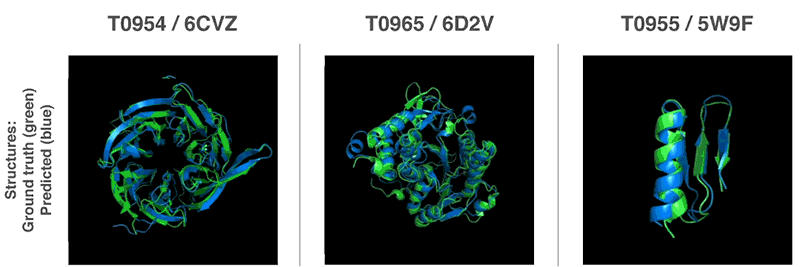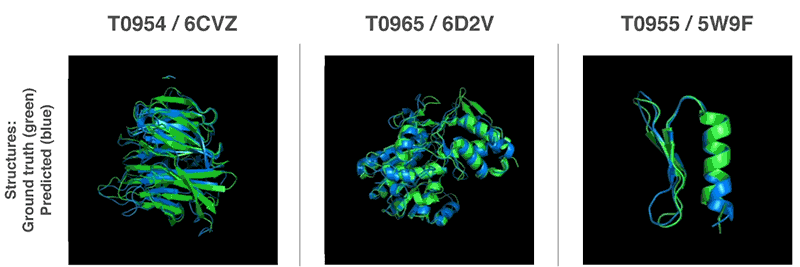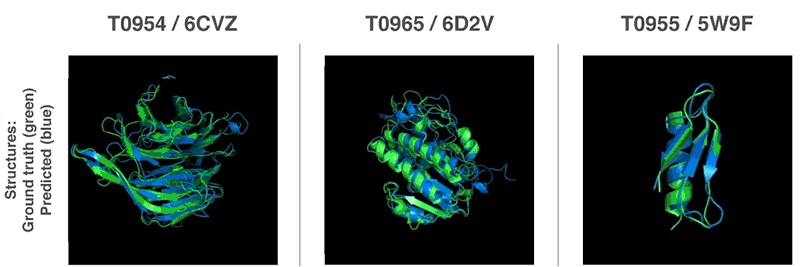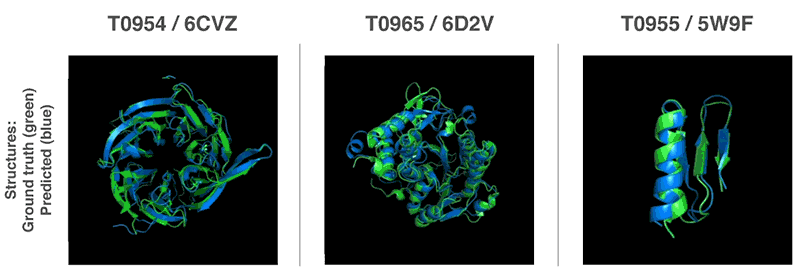
Источник: DeepMind
Понимание структуры белков определенного типа позволит создать реагенты, которые смогут активно воздействовать на эти белки. В качестве юзкейса можно назвать ликвидацию разлившейся нефти или создание недорогого быстро разлагающегося пластика.
По словам одного из представителей DeepMind, их исследование является предвестником новой эры. Работа относится к числу тех, что решают фундаментальные проблемы как науки, так и технологий. Стоит отметить, что специалисты DeepMind занялись созданием нового ИИ после того, как их алгоритм AlphaGo победил в игре в го Ли Седоля, чемпиона мира.
После этого ИИ научили проходить сложные для машин компьютерные игры, включая «Месть Монтесумы». Разработчики говорят, что их целью никогда не было получить побольше очков в какой-либо игре, чтобы показать силу своего ИИ. Настоящая цель — разработка алгоритмов, которые могут помочь человеку в решении вопросов науки и техники, таких, как структура белков и ее предсказание.
Ученые смогли научить AlphaFold определять расстояние между парами аминокислот, а также конфигурацию химической связи. Второй этап заключался в поиске наиболее энергоэффективной структуры каждого предполагаемого белка. Сейчас у алгоритма на выполнение задачи уходит всего несколько часов — в то время, как люди тратят на то же самое месяцы или даже годы.
Комментарии (43)

Tamul
04.12.2018 01:10-1Почитал про AlphaFold, потыкал по ссылкам, но так и не понял, чем их алгоритм на глубоких нейронных сетях и машинном обучении (опять?) круче алгоритмов, годами крутящихся на компьютерах энтузиастов в Rosetta@home.
perfect_genius
04.12.2018 08:22Тем, что участвовала в конкурсе и выиграл, а Rosetta@home — нет?
vesper-bot
04.12.2018 10:28Розетта — не нейронная сеть, чтобы иметь возможность участвовать в конкурсе. А перепилить её под нейронные сети может оказаться и невозможным.
krioz
04.12.2018 12:34тем, что после обучения AlphaFold за неделю высчитает то, что Rosetta высчитывает годами?
KvanTTT
04.12.2018 13:07Хм, а можно ли будет в каком-то виде встроить этот алгоритм в BOINC и распараллелить на все пользовательские компы (по аналогии с Rosetta)?
krioz
04.12.2018 13:24Скорей всего нет, т.к. DeepMind использует собственные TPU (Tensor Processing Unit) для вычислений(вроде как).
Если же вы подразумевали, что Rosetta возьмет верх над AlphaFold за счет кол-ва пользователей, то здесь можно вспомнить, что самая мощная версия AlphaGo(Zero) использовала одну машину с всего 4-мя TPU v2 для своих вычислений.KvanTTT
04.12.2018 18:02Ну я и написал «в каком-то виде», т.е. если портировать алгоритмы на GPU например.
crazy_llama
04.12.2018 13:29Скорее всего это смесь классических методов и нейронной сети. Как тривиальный пример: осуществляешь обычный перебор, но начинаешь с того, что тебе советует НС. У них похожий концепт был в AlphaGo.
KvanTTT
04.12.2018 18:04Там используется методика Монте-Карло с проигрыванием большого количества случайных партий. Думаю что с фелками подобное возможно: генерация случайных свернутых белков.
DaylightIsBurning
04.12.2018 18:13а откуда узнать структуру белка случайной последовательности? Число известных структур белков очень ограничено (менее 150 тысяч, не все записи там уникальные белки). Более того, есть целые группы белков, где мы не знаем почти ничего. Лишь для 7 организмов известно более 500 структур белков. Когда в CASP появляется target из «экзотического» для PDB организма (то есть не из топ 10 исследованных организмов или их близких эволюционных родственников), он может не поддаваться существующим методам совершенно, даже приблизительно. Недавно был пример какого-то белка из утконоса — даже близко не смогли компьютерным моделированием найти структуру.
vershinin
04.12.2018 23:55Да, так и есть. Скорей всего Альфафолд смог обнаружить гомологичные белки и не увидел уникальные. С учетом того, что некоторые белки организуют соединения с металлами, с углеводами, сворачиваются только в присутствии других белков или при определенных условиях (pH, температура, и так далее), подвергаются посттрансляционной обработке, то задача становится очень сложной. Даже одна заменённая аминокислота может повести белок совсем по другому пути сворачивания. С другой стороны, это большое достижение — доказывает, что там есть какой-то паттерн, механизм или код, который было бы неплохо изучить и понять. Количество уникальных белков тоже постепенно сокращается, так что точность будет только расти. Да и вообще, в конструировании новых белок этого задела должно вполне хватить. Другое дело, что одним фолдом тут не отделаешься, нужно знать как выглядит активный центр, а его на NMR и кристаллографии не видно, потому что активные группы постоянно находятся в движении. В общем, достижение большое, но недостаточное для переворота в области.
DaylightIsBurning
05.12.2018 02:56Не, они фокусировались именно на free modelling targets.
Их алгоритм построен не на шаблонах, а на coevolution подходе, который стал модным в последние 4-5 лет, хотя идея была впервые озвучена еще в 1990е. Они описывают это в своем блоге.
Теоретически возможно, что AlphaFold лучше ищёт шаблоны, чем остальные участники и, по сути, для него больший процент целей является template. Уже были одиночные случаи, что один из участников находил шаблон, которые другие отбрасывали и получал уникально хорошее решение. Но, как я понимаю, AlphaFold не использует шаблоны ни в каком виде, так что дело, видимо, не в этом.vershinin
05.12.2018 10:08+2На сколько я понимаю, вы не плохо разбитаетесь в теме. В своё студенческое время я тоже пытался заниматься de novo prediction, но потом соскочил в другую область. У с того времени остались вопросы, может быть вы попробуете на них ответить, или тот ваш товарищ, который на CASP ездил.
- Насколько верна догма Анфинсена? Как я понимаю, чтобы её доказать, он разрушал дисульфидные мостики в белке, белок терял активность, затем дисульфидные мостики восстанавливал — и белок снова начинал работать. Есть большая вероятность, что белок был восстановлен шаперонами, или был разложен не до конца. Были ли поставлены эксперименты, где белок синтезировали с нуля химическим способом и он свернулся в биологически активную форму?
- Почему при de novo предсказаниях не используется биологический подход, ведь белок выдавливается из рибосомы по одной аминокислоте в «водяной мешок» и эти аминокислоты начинают взаимодействовать друг с другом не сразу все, а по очереди? Когда белки восстанавливаются при помощи шаперонов, они тоже проходят через «трубку», и сворачивание происходит с учётом последовательности. Так что может быть парадокс Левенталя не такой и парадокс?
- То, что белок начинает сворачиваться ещё до того, как сошёл с рибосомы уже доказанный факт. Пост-трансляционная обработка может изменить структуру, но очень незначительно, что заставляет нас опять смотреть в сторону трансляции для предсказания структуры. Есть доказанный экспериментом факт, что если в последовательности заменить одну аминокислоту, то мы можем получим совсем другую укладку- вместо нескольких бета-спиралей глобулярного белка получаются альфа-листы. То, что эта аминокислота находилась в середине последовательности открывает простор для некоторых мыслей, но это уже отдельный разговор.
- Если натравливать нейронные сети, то может быть стоит их использовать таким образом, как их натравливают на распознавание человеческой речи? А не давать на вход всю последовательность, надеясь что нейронная сеть увидит какие-то свои шаблоны.
Заранее спасибо, если сможете успокоить моё любопытство.DaylightIsBurning
05.12.2018 12:27Насколько верна догма Анфинсена
Догма была сформулирована для «small globular protein» и в основном верна, но есть исключения. Есть пост-трансляционные модификации. Прионы тоже в некотором смысле можно считать исключением, но можно не считать, а отнести к «неверным» условиям внешней среды. То же самое с лигандами типа ионов, сахаров и т.п.
Были ли поставлены эксперименты, где белок синтезировали с нуля химическим способом и он свернулся в биологически активную форму?
Были, свернулся, можно сначала поместить белок в среду с unfolding agent, например мочевину, затем снизить концентрацию мочевины и белок успешно сворачивается назад. Но гарантий, то все белки так могут нет. Скорее всего даже уже были примеры, что некоторые не могут, но я не следил.
Почему при de novo предсказаниях не используется биологический подход
Используется, rosetta, например по кускам собирает. Не думаю что это критически важно. Проблема с этим подходом в том, что он подвержен локальным минимумам, которые в природе не проблема — времени вагон что бы исправить фолд, а в симуляции — нет. Не факт что последовательный выход — необходимость. Вероятно, пока весь белок не вылезет он всё равно нормально не свернется, то есть последовательные выход из туннеля рибосомы может не быть критичен для сворачивания. Тем более для компьютерного определения структуры — процесс свертки можно пропустить, вероятно, хотя, возможно, не всегда.
может быть парадокс Левенталя не такой и парадокс?
Этот парадокс разрешен через folding funnel. Нет необходимости привлекать ещё и объяснение через процесс сворачивание по одной аминокислоте при синтезе.
То, что белок начинает сворачиваться ещё до того, как сошёл с рибосомы уже доказанный факт.
Тут и доказательство не требуется, как иначе-то? Вопрос только в том, имеет ли это значение? Доказано, что, как минимум, не всегда. Кроме того, не обязательно повторять естественный путь сворачивания в компьютере, нас не путь а конечная структура интересует, которая независимо от пути термодинамически/кинетически стабильна. Что лучше работает — то и используют. Сейчас лучше получается сворачивать используя свойства свернутого состояния, а не пути сворачивания. Поиск пути сворачивания может быть более сложной и менее интересной (полезной) задачей. А может и нет, но пока так :).
может быть стоит их использовать таким образом, как их натравливают на распознавание человеческой речи?
Если Вы имеете ввиду «по кускам», то этот подход активно применяется и применялся. Та же rosetta, сшивание (под-)шаблонов и многие многие другие.

LinearLeopard
05.12.2018 09:28Почитал про AlphaFold, потыкал по ссылкам, но так и не понял, чем их алгоритм на глубоких нейронных сетях и машинном обучении (опять?) круче алгоритмов, годами крутящихся на компьютерах энтузиастов в Rosetta@home.
Я вообще не нашёл их в конкурсе, хотя может это я такой слепень, кто-нибудь подскажите, пожалуйста, финальную таблицу, если нашли.
leshabirukov
04.12.2018 16:15Интересно, обратную задачу смогут? Есть, к примеру, состязательные сети для такого. Или уж сразу по требуемым свойствам выдавать технологию синтеза, или хотя бы ДНК-последовательность. Вот будет ГМО так ГМО.
DaylightIsBurning
04.12.2018 16:54Эта область науки называется protein design, и группа David Baker активно и давно работает в этом направлении. Эта же группа является одной из самых успешных участников CASP многие годы (постоянно входит в топ, в некоторых категориях — топ 1).
leshabirukov
04.12.2018 18:32А насколько именно техника Deep Mind новая, до них нейросети успешно применяли?
Abiron
04.12.2018 18:34Применяли для белков или вообще?
Тут очень важна область применения, разные структуры нейросетей сильно по разному хороши в решении разных типов задач.
Сами сети глубокого обучения стали популярны не так давно, а для подобных задач вроде игры в GO или вычисления белка применяются еще меньше по времени.
Если ваш вопрос про то можно ли ожидать взрывного роста отдельных областей за счет применения нейросетей (я его понял так) — вполне, сейчас нейросети новых структур эффективно применяют для задач где еще 10 лет назад даже и не рассматривались.leshabirukov
04.12.2018 18:43Под «вообще» вы имеете в виду вывести свойства вещества по формуле или наоборот, а также поиск способа синтеза? Ну да, и про это и про белки интересно было бы послушать, думаю и статья пользовалась бы успехом тут.
DaylightIsBurning
04.12.2018 18:49для белков способ синтеза искать не надо, технологии позволяют синтезировать любой выдуманный белок заданный последовательностью аминокислот. Сложность в том что бы предсказать трехмерную структуру и функцию белка по его формуле.
leshabirukov
04.12.2018 19:19Понятно, (хотя можно позанудствовать про дополнительную модификацию или шапероны, очень возможно, что с развитием технологии станет актуально). Ну и я так понимаю, ощущения, что DM по привычке всех порвет пока нет.
DaylightIsBurning
04.12.2018 19:43+1Через пару дней мой коллега должен вернуться с CASP конференции — расспрошу, какие у него впечатления по поводу AlphaFold, но пока мне кажется, что тут скорее речь про значительное инкрементальное, но не радикальное улучшение state of the art.
DaylightIsBurning
04.12.2018 18:43+1Могу точно сказать, что нейросети для определения структуры белков применяли и успешно, но не факт, что таким же способом как Google, не знаю как именно работает AlphaFold.
DaylightIsBurning
04.12.2018 20:16+1Прочитал их пресс-релиз, и если я его правильно понял, они реализовали появившуюся лет 5 назад технологию co-evolution based contact & structure prediction. В общих чертах подход известный. Видимо, их опыт с нейронками и огромные вычислительные ресурсы позволили им реализовать количественно более точную модель чем у конкурентов из университетов.
Уже были попытки (успешные) реализовать схему аналогичную той, что они показывают:

Из любопытного — они натренировали нейронку для оценки неправильности модели. Использование статистического потенциала для этих целей — старый метод, не знаю, пробовал ли кто-то раньше нейронки для этого.
We also trained a separate neural network that uses all distances in aggregate to estimate how close the proposed structure is to the right answer.
Что именно обеспечило преимущество мне пока неясно — просто бОльшие вычислительные ресурсы или вот эта идея с усложнением (улучшением) scoring function до нейронки или ещё что-то.leshabirukov
05.12.2018 11:06Про дополнительную сеть, — вот и черты соревновательных сетей появляются. Следующий шаг, — не только мерять «неправильность модели» по «distances in aggregate», а тренировать настоящий дискриминатор реальных белков от предсказанных сетью.
DaylightIsBurning
05.12.2018 12:33Не понял про соревновательность, где она тут? Тренировка сетей на дискриминацию реальных белков усложнена малым числом известных положительных примеров — всего 150 тысяч. Положительные примеры приходится определять экспериментально — долго, дорого.
Вообще измерить неправильность модели по известной contact map легко, есть даже функции неправильности, которые вообще без ничего (без contact map) работают, Rosetta scores. То есть понять, что полученная модель правильная — не очень сложно, если она есть. Проблема в том, как получить гладкую функцию неправильности, когда модель ещё не совсем близка к верной, иначе градиентный спуск не работает (именно его применяет AlphaFold). Их нейронка, похоже, именно такую функцию неправильности дала.
Я не знаю, как именно они это делали, но мне на ум пришел следующий алгоритм. Можно попробовать натренировать нейронку на оценку неправильности в «числе изменений». Берем реальную структуру — неправильность = 0, меняем один элемент (заменяем фрагмент или dihedral) — неправильность = 1, возвращаем назад, меняем другой элемент — тоже неправильность = 1, далее меняем два подряд — неправильность = 2 и т.д. На этом ансамбле фальшивых моделей и соответствующих им метрик неправильности можно, наверное, натренировать нейронку.leshabirukov
05.12.2018 12:58Ну, берем основную сеть в качестве генератора, а дополнительную, — дискриминатора. Когда дополнительная оценивает результат, она фактически штрафует основную за неправдоподобие… Хотя наверное я тут неправ, поскольку генератор имеет доступ к образцам, и на входе имеет не шум, а первичную структуру. Но всё же предположу, что ситуации, когда одна сеть используется для вычисления ошибки другой, можно рассматривать как обобщение соревновательных сетей.
DaylightIsBurning
05.12.2018 13:35Мне кажется, если основная и дополнительная сети тренируются на одних данных и отличаются только тем, что одна генераторная, а вторая — дискриминатор — это по сути одна сеть. Генератор сам себя штрафует за неправдоподобие, зачем второй раз?
leshabirukov
05.12.2018 16:54Дополнительная ничего не знает о входе (первичной структуре) и специализируется на оценке правдоподобия. Она просто помогает основной учиться.
DaylightIsBurning
05.12.2018 17:33а на чем будем учить эту сеть? На известных структурах? Можно, так делают в виде стат. потенциала это есть в Rosetta. Есть соблазн превратить стат. потенциал в нейронку, но есть две проблемы: всего 150000 моделей доступно для тренировки и скорость работы. Посчитать стат потенциал — быстро, оценить scrore через нейронку — не знаю, так ли быстро.
leshabirukov
05.12.2018 17:57А расширение данных к этим 150000 моделям не применимо ли? Просто повертеть в пространстве, как с картинками обычно делают.
DaylightIsBurning
05.12.2018 18:18ну в этом особо нет смысла, эти данные можно представить в виде пар расстояний и тогда все повороты уже учтены выходит. Или можно центрировать координаты на центр масс и ориентировать по тензору инерции. Короче нет проблем уникальное представление получить. В картинках повороты нужны что бы объяснить сети, что повороты — одно и то же. В случае белков входные картинки не имеют поворотов, этой проблемы не существует.
leshabirukov
05.12.2018 22:03В MNIST-е цифры тоже отцентрованы, тем не менее расширение путем параллельного переноса успешно применяли (https://ru.wikipedia.org/wiki/MNIST, примечания 14, 15, 16).
DaylightIsBurning
04.12.2018 17:48+1А ещё, уже довольно давно химическая промышленность применяет искусственно-модифицированные белки-катализаторы (энзимы). Обычно эти белки создаются так: берется известные белок с нужной функцией из какого-нибудь организма (например млекопитающего), ищут его аналоги в других организмах (например, полярной рыбы), определяют механизм действия, различия в эффективности в зависимости от условий реакции (температура, pH и т.д.). А затем искусственно целенаправленно или наобум меняют понемногу формулу белка (ГМО) пока не получится энзим с более подходящими для промышленности свойствами.
Потом синтезируют ДНК с вновь полученной формулой и подсаживают эту ДНК каким-нибудь бактериям (например), кормят эти бактерии на убой, бактерии копируют подсаженную «паразитную» ДНК, производят на её основе белок. Затем бактерий убивают, разрушают ультразвуком, из полученной каши и отфильтровывают нужный белок.
DaylightIsBurning
04.12.2018 17:10ИИ удалось правильно предсказать структуру 25 из 43 белков. На втором месте — команда, которой удалось правильно предсказать структуру 3 из 43 белков
Фактическая ошибка, дает совершенно ошибочное представление о случившемся. На самом деле AlphaFold удалось предложить САМУЮ лучшую модель среди всех участников для 25 из 43 белков. То есть на самом деле в 25 случаях из 43 AlphaFold дали ответ чуть-чуть точнее чем конкуренты. Загадали 43 задачи, AlphaFold дала 25 ответов, каждый из которых верен с точностью 1 ангстрем (условно), а команда, которая заняла второе место на те же 25 задач дала ответы верные с точностью 1.01 ангстрема. Разница совершенно небольшая. На самом деле в CASP не эта метрика главная, а, грубо говоря, число достаточно верно угаданных структур. По этой метрике AlphaFold не так сильно лидирует (120.3 балла против 107.0 у второго места). Нет особо смысла уточнять цифры после 3го знака для «простых» задач, а именно на этом фокус в статье.
И ещё одна ошибка:
В организме человека содержится огромное количество разновидностей белка. По разным оценкам, оно может достигать нескольких миллиардов
Открываем статью и что видим:
...there should be at least ~20,000 nonmodified (canonical) human proteins. Taking into account products of alternative splicing (AS),… as many as 100 different proteins can potentially be produced from a single gene
Итого 2 миллиона, а не «несколько миллиардов».LinearLeopard
05.12.2018 09:31DaylightIsBurning
А можно попросить вас указать ссылку на финальные результаты, а то я совсем не в теме, так и не смог разобраться, где же они на сайте, заранее спасибо.DaylightIsBurning
05.12.2018 10:56+1predictioncenter.org/casp13/zscores_final.cgi
там нужно отметить Targets: TBM/FM, FM
ещё есть Table Browser — можно там смотреть, но ссылки дать не могу — оно всё POSTом настраивается. Google — это A7D.
DaylightIsBurning
05.12.2018 18:40Посмотрел на результаты, вот для тех кому лень разбираться с сайтом. Сравнение AlphaFold и Zhang(второе место в этом году). GDT_TS — это метрика правильности ответа. Больше — лучше: 100 — идеальное предсказание, 20 — случайный левый белок. Разница между GDT_TS 70 и 72, к примеру, обычно не слишком значительная — это модели сопоставимого качества.
| # | Model | GDT_TS A7D | GDT_TS Zhang | Winner | GDT_TS diff | GDT_TS best | |----|--------------|------------|--------------|--------|-------------|-------------| | 1 | T0955_1-D1 | 87.81 | 85.98 | A7D | -1.83 | 87.81 | | 2 | T0990_1-D1 | 85.2 | 64.8 | A7D | -20.40 | 85.2 | | 3 | T1008_1-D1 | 81.49 | 40.58 | A7D | -40.91 | 81.49 | | 4 | T0968s2_1-D1 | 78.7 | 62.39 | A7D | -16.31 | 78.7 | | 5 | T1019s1_1-D1 | 78.02 | 71.55 | A7D | -6.47 | 78.02 | | 6 | T0992_1-D1 | 75.94 | 73.6 | A7D | -2.34 | 75.94 | | 7 | T0997_1-D1 | 74.86 | 55 | A7D | -19.86 | 74.86 | | 8 | T1015s1_1-D1 | 72.44 | 58.52 | A7D | -13.92 | 72.44 | | 9 | T0958_1-D1 | 71.43 | 62.99 | A7D | -8.44 | 71.43 | | 10 | T1017s2_1-D1 | 71.2 | 69 | A7D | -2.20 | 71.2 | | 11 | T0968s1_1-D1 | 70.13 | 56.78 | A7D | -13.35 | 70.13 | | 12 | T0986s2_1-D1 | 70 | 46.77 | A7D | -23.23 | 70 | | 13 | T0986s1_1-D1 | 65.76 | 69.02 | Zhang | 3.26 | 69.02 | | 14 | T1000_1-D2 | 68.75 | 61.21 | A7D | -7.54 | 68.75 | | 15 | T0953s2_1-D2 | 56.31 | 65.54 | Zhang | 9.23 | 65.54 | | 16 | T1021s3_1-D2 | 65.46 | 49.23 | A7D | -16.23 | 65.46 | | 17 | T0987_1-D1 | 63.65 | 48.92 | A7D | -14.73 | 63.65 | | 18 | T0970_1-D1 | 63.23 | 58.82 | A7D | -4.41 | 63.23 | | 19 | T1001_1-D1 | 62.23 | 59.89 | A7D | -2.34 | 62.23 | | 20 | T0949_1-D1 | 61.63 | 62.21 | Zhang | 0.58 | 62.21 | | 21 | T1021s3_1-D1 | 61.6 | 50.45 | A7D | -11.15 | 61.6 | | 22 | T0963_1-D2 | 42.07 | 61.59 | Zhang | 19.52 | 61.59 | | 23 | T0957s2_1-D1 | 60.48 | 54.52 | A7D | -5.96 | 60.48 | | 24 | T0975_1-D1 | 58.9 | 42.79 | A7D | -16.11 | 58.9 | | 25 | T0960_1-D2 | 39.88 | 58.63 | Zhang | 18.75 | 58.63 | | 26 | T1005_1-D1 | 56.37 | 51.38 | A7D | -4.99 | 56.37 | | 27 | T0981_1-D3 | 55.17 | 53.33 | A7D | -1.84 | 55.17 | | 28 | T0957s1_1-D1 | 54.86 | 42.13 | A7D | -12.73 | 54.86 | | 29 | T0953s1_1-D1 | 54.48 | 41.05 | A7D | -13.43 | 54.48 | | 30 | T0989_1-D1 | 41.42 | 53.92 | Zhang | 12.50 | 53.92 | | 31 | T1022s1_1-D1 | 50.64 | 52.4 | Zhang | 1.76 | 52.4 | | 32 | T0987_1-D2 | 52.04 | 35.97 | A7D | -16.07 | 52.04 | | 33 | T0969_1-D1 | 51.7 | 48.3 | A7D | -3.40 | 51.7 | | 34 | T0953s2_1-D1 | 51.14 | 50 | A7D | -1.14 | 51.14 | | 35 | T0990_1-D3 | 48.71 | 16.78 | A7D | -31.93 | 48.71 | | 36 | T0990_1-D2 | 45.89 | 29.76 | A7D | -16.13 | 45.89 | | 37 | T0953s2_1-D3 | 29.3 | 43.01 | Zhang | 13.71 | 43.01 | | 38 | T0980s1_1-D1 | 41.83 | 42.55 | Zhang | 0.72 | 42.55 | | 39 | T0989_1-D2 | 38.84 | 32.14 | A7D | -6.70 | 38.84 | | 40 | T1010_1-D1 | 29.64 | 33.33 | Zhang | 3.69 | 33.33 | | 41 | T0981_1-D2 | 24.06 | 31.56 | Zhang | 7.50 | 31.56 | | 42 | T0991_1-D1 | 25.9 | 23.42 | A7D | -2.48 | 25.9 | | 43 | T0998_1-D1 | 18.68 | 17.02 | A7D | -1.66 | 18.68 |vershinin
05.12.2018 21:55Хорошо, но у Zhang высококвалифицированные спецы, а у всего лишь A7D нейронная сеть? Если это так, то это должно быть большое достижение же!
perfect_genius
Не знал про количество видов белков, наше тело крайне сложное. Учитывая разнообразие его органов-систем, скорее всего, в мозге намного меньше разных типов белков, чем в остальном теле.
DaylightIsBurning
Да это «ученый изнасиловал журналиста». На самом деле в геноме белков с разными последовательностями аминокислот всего лишь ~20000, с учетом пост-трансляционных модификаций и т.п. выходит максимум несколько миллионов «подвидов», из которых большинство — потенциально возможные модификации одних и тех же белков, которые, вероятно не имеют значения. Не имеют значения в том смысле, что это случайные изменения, которые не меняют функцию и свойства белка а просто случайные безобидные «деффекты».