
Введение
О чём этот текст
Если человек услышит о «симуляции реальности», то в наиболее вероятно ему в голову придут или разные научно-фантастические произведения (типа Матрицы, Темного города, или Теоремы Зеро), или компьютерные игры. В случае людей, чьи головы засорены инженерным образованием, возможно всплывут пакеты типа
А ведь есть ещё один уровень Реальности, который окажется незаслуженно забытым: тот, на котором происходит вся химия — это уровень атомов и молекул. Его тоже можно вполне успешно моделировать на компьютере. Поскольку в данном срезе Реальности всем заведует квантовая механика, то подобные расчёты часто называют квантовой химией. И вот о её связи с Реальностью, изучаемой экспериментальными методами, мы и поговорим.
Этот текст будет об элементарнейших вещах. Но, практика чтения научных журналов и слушания различных докладов показывает, что об этом надо постоянно напоминать.
Текст рассчитан на людей понимающих и/или интересующихся тем, как живут атомы и молекулки.
Взято из xkcd.com
Физические методы исследования жизни молекул
Мы знаем из школьных курсов химии и физики, что все вещества состоят из атомов, молекул, ионов, или их комбинаций. И мы, вроде, даже знаем какой жизнью они живут. Но у этой инфы должны быть свои надёжные источники (методы исследования), и они действительно есть.
Таких способов подсмотреть за жизнью атомов существует очень и очень много. Желающие, например, могут ознакомиться с некоторыми из них поподробнее в классических учебниках
— Пентин Ю.А., Вилков Л.В. Физические методы исследования в химии. — М.: Мир, 2006,
— Драго Р. Физические методы в химии. — М.: Мир, 1981.
Но, грубо и достаточно легко выделяются 3 основные группы методов:
- спектроскопические методы,
- дифракционные методы,
- различные методы микроскопии (не важно, просвечивающей или сканирующей, для нас это сейчас не существенно).
Про последнюю речи не будет, но её инструментарий не менее важен, чем у первых двух.
Спектроскопические методы исследования вещества
Эта мощная группа методов обеспечивает нас очень и очень многим: от поиска и определения молекул в межзвёздной среде и на других планетах до банальной проверки на взрывчатые вещества в аэропорту.
Общий принцип работы спектральных методов
Когда говорят о спектроскопии, обычно имеется в виду следующий общий принцип работы.
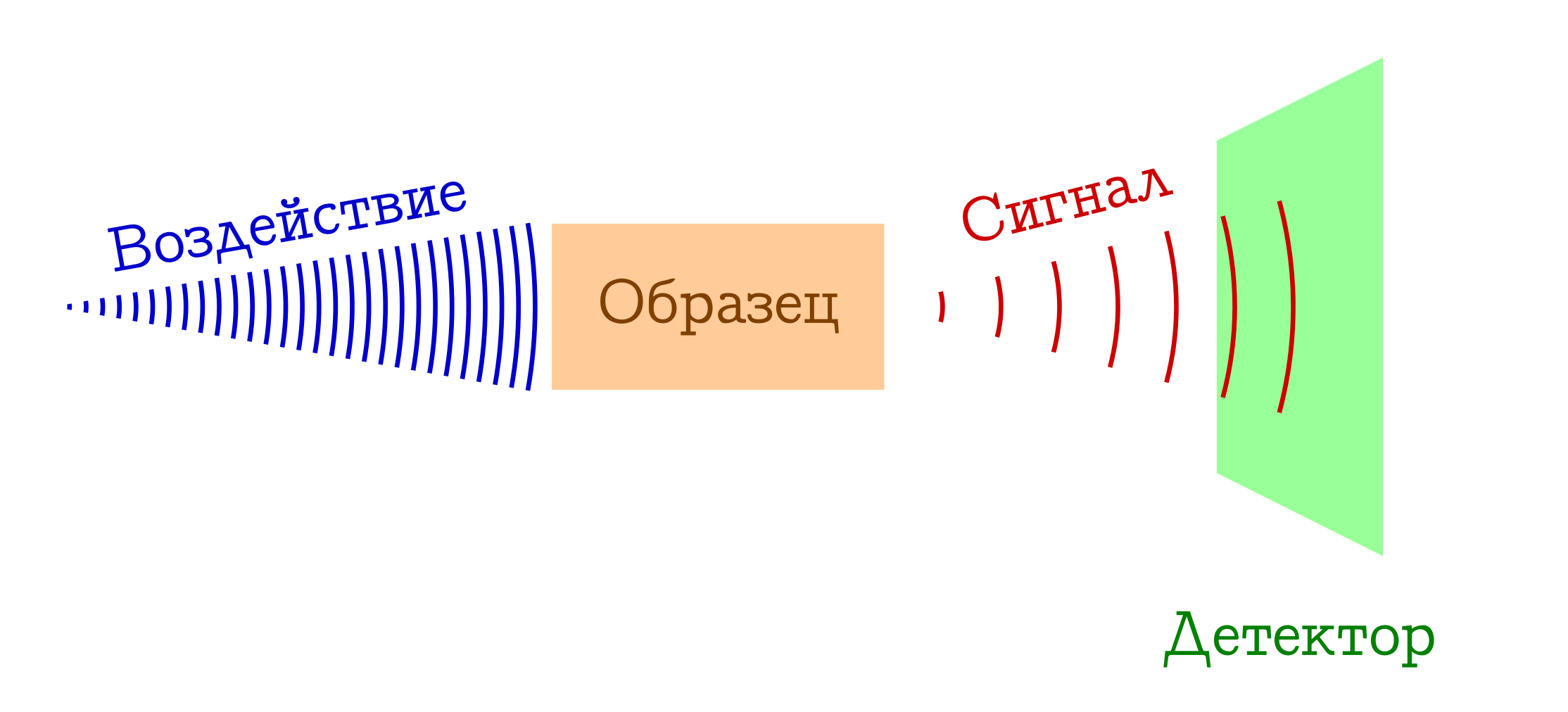
Общая схема спектральных методов исследования вещества
- У нас есть нечто, чем мы (например, лампочкой/лазером/солнечным светом) воздействуем на интересующий нас образец. Чаще всего это электромагнитное изучение, но этим вполне могут быть электроны (например, в масс-спектроскопии с ионизацией электронным ударом) или коктейль всего возможного и невозможного из плазмы (например, в спектроскопии пламён, так любимой школьниками и студентами младших курсов химфаков). Так или иначе, на наш образец что-то должно подействовать.
- С образцом при воздействии происходит нечто такое, что он меняет своё состояние. Это может быть переход на какой-то возбуждённый уровень (в любой спектрофотометрии или спектроскопии комбинационного рассеяния), или вообще развал молекулярной системы (как в масс-спектрах или фотоэлектронной спектроскопии). Но так или иначе образец в какой-то момент должен быть другим.
- ???
- PROFIT!!! Мы регистрируем некий сигнал (испущенный или поглощённый) при этом изменении образца на молекулярном уровне. Это могут быть потерянные фотоны, затраченные на изменение образца (тогда имеем абсорбционную спектроскопию), или же наоборот, лишние фотоны, испущенные после предварительного возбуждения вещества (эмиссионную спектроскопию), изменение длины волны изначальных фотонов в результате взаимодействия с веществом (спектроскопия комбинационного рассеяния, более известная за рубежом как
раменовскаярамановская), ну или тупо осколки изначальных молекул (как в масс-спектрах или фотоэлектронной спектрскопии). Вариантов много — суть одна: сигнал есть!
В качестве примера подобных методов можно привести кучу разных буквочек: NMR, ESR, MW, THz, IR, UV/Vis, XRF, MS, PES, EXAFS, XANES и т.д. и т.п.
Все (или многие из них) знакомы (или должны быть знакомы) каждому химику. Все эти методы — это (далеко неполный) стандартный арсенал уважающего себя исследователя, имеющего дело с веществами.
Спектральные диапазоны и их связь с жизнью молекул
Взято из xkcd.com
Поскольку в подавляющем числе случаев спектроскопия всё же завязана на электромагнитное излучение, то логично привязать диапазоны электромагнитного спектра к различным аспектам атомно-молекулярной жизни. Ведь частота используемых в спектроскопии электромагнитных волн является своего рода «часами», позволяющими засечь, сколько длится тот или иной процесс в молекулярных системах. А значит, меняя эту частоту можно изучать (и даже воздействовать) на разные молекулярные процессы.
Итак.
- В сверхдлинноволновом диапазоне с химической точки зрения ничего интересного не происходит, так что о нём можно не вспоминать.
- С частотой радио- и микроволн (и даже длинноволнового инфракрасного излучения, ИК=IR) вращаются разные молекулы в газовой фазе: большие и тяжёлые — в области радиоволн (более низкие частоты), а маленькие и легкие — в ИК (более высокие частоты).
- В ИК же (в основном) происходят различные колебания молекул: всякие конформационные и другие не очевидные движения внутри молекул — в длинноволновом ИК, а валентные колебания (растяжения — сокращения длин химических связей) — в коротковолновом (до 4000 см–1).
- Ну а дальше идёт место спектра, где обитают разные электронные переходы (вплоть до области ?-квантов). При меньших частотах (видимый, УФ=UV и мягкий рентген) живут в основном переходы, связанные с валентными электронами.
Почему мы видим?Кстати, именно, из-за электронных переходов мы и можем видеть: в наших глазах (в колбочках) содержатся структуры, имеющие в составе ретиналь. При поглощении фотона видимого цвета этой молекулой, в ней происходит разрыв двойной связи, что приводит к цис-транс изомеризации. И именно это изменение воспринимается нами как первичный сигнал, который далее передаётся в наш мозг.
Но при повышении энергии фотонов (т.е. при повышении частоты, как мы помним по формуле Планка ) мы добираемся до всё более и более глубоких слоёв электронной структуры, пока не упираемся в рентгеновском диапазоне до финальных 1s-оболочек (или, как их зовут рентгенщики, K).
Так вот, выбирая правильную длину волны электромагнитного излучения, мы можем посмотреть подробнее на тот или иной процесс в молекулах.
Дифракционные методы исследования вещества
Теперь немного поговорим о дифракции. Принципиальная схема подобных экспериментов тоже проста.
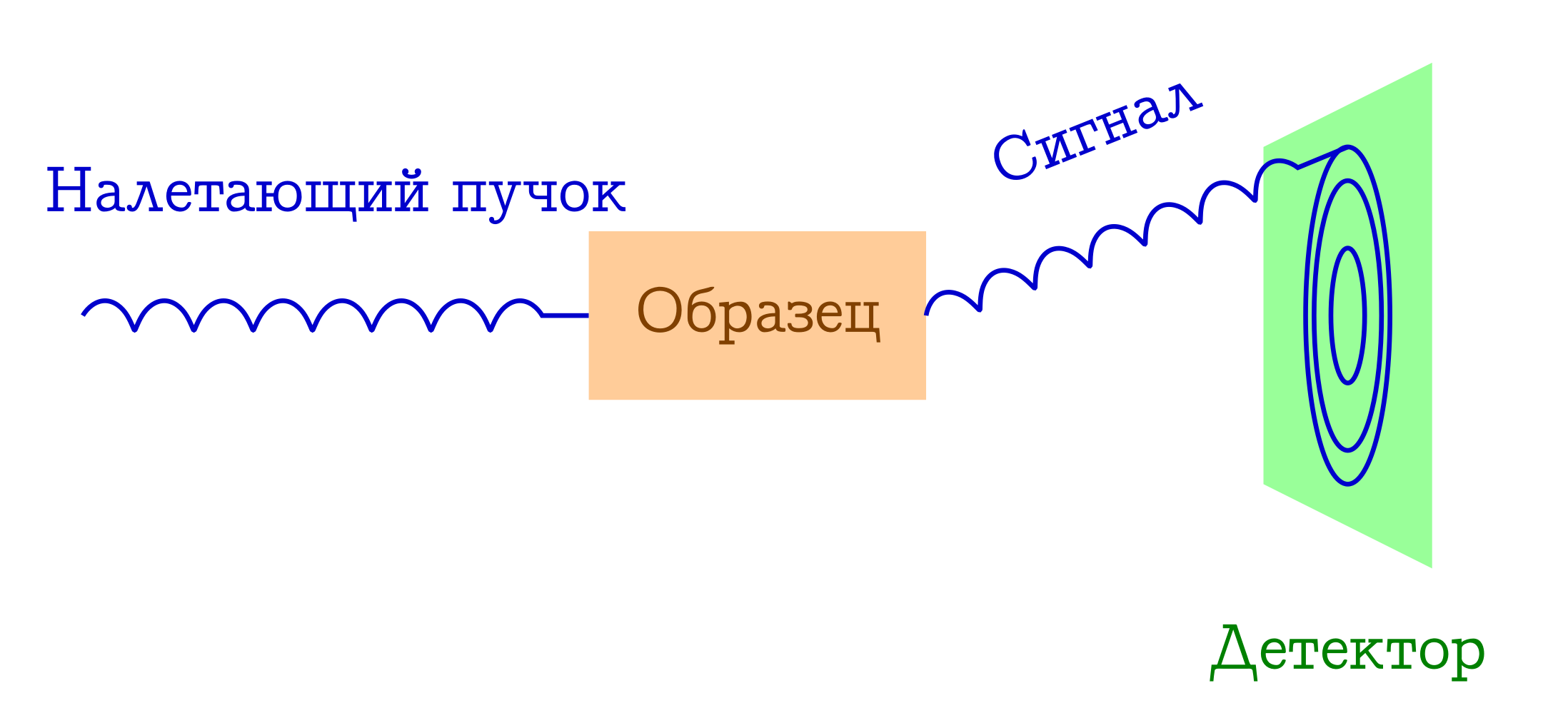
Общая схема дифракционных методов исследования вещества
- На образец налетает пучок каких-то частиц. Чаще всего это или рентгеновские фотоны, или электроны, или нейтроны.
- Эти частицы по разным механизмам упруго рассеиваются на атомах в интересующем нас образце (т.е. без изменения длины и фазы волны, просто меняют направление своего полёта). С самим образцом от этих налетевших частиц ничего не происходит: он просто не успевает на них среагировать.
- Межатомные расстояния служат в качестве дифракционной решётки для налетевшего пучка, поэтому в результате на детекторе мы увидим красивую дифракционную картинку.
Из последнего пункта появляется условие на длину волны налетающих частиц (?): она должна быть того же порядка или меньше, чем характеристический порядок межатомных расстояний, поэтому типичные ? для данных методов составляют 1 — 0.01 A.
Основные типы ошибок при сравнении экспериментов и теоретических расчётов
В итоге мы имеем очень интересную картину: и в спектроскопии и в дифракции мы наблюдаем какой-то левый сигнал, который как-то косвенно свидетельствует о том, что происходит в молекулярной системе на самом деле.
Но, по счастью, иногда мы можем теоретически рассчитать интересующий нас сигнал (как, например, в микроволновой, ИК или UV/Vis спектроскопии), а иногда мы можем из наблюдаемого сигнала извлечь интересующие нас величины, доступные для квантово-химического расчёта (например, расстояния между атомами в молекуле, дипольный момент и т.д.). И тут у нас возникает шанс, что численный и настоящий эксперимент могут объединиться в страстной стадии сравнения друг с другом… и здесь стандартно могут возникать 4 типа ошибок.
Внимание! Термин «ошибка» здесь означает не то, что результат сравнения заведомо получается неправильный. Просто почва для сравнения становится очень зыбкой и болотистой, и один неаккуратный шаг может легко запороть всю работу.
- Разные условия эксперимента и/или расчёта (агрегатное состояние, температуры, давления и т.д.). Мы можем внезапно начать сравнивать между собой разные системы, по какой-то причине считая их одинаковыми. Например очевидно, что добавление одной или пяти чайных ложек сахара к чашке чая приведёт к одной и той же физической системе под названием «чай с сахаром», но свойства у этой системы будут очень разные. И это можно легко измерить. Например, термометром (замерив температуру чая сразу после растворения сахара) или языком (одним из т.н. органолептических методов анализа). Так что сравнивая полученные системы между собой (будь это реальная чашка сахара с чаем, или её компьютерная модель) надо не забывать, что сходство имеет свои границы, и что если мы уменьшим допустимую погрешность для «схожести», то в итоге отыщем различия.
- Разный физический и/или математический смысл параметров (у параметра физического смысла в привычном смысле может вообще не быть). Тут тоже всё просто: если мы сравниваем 2 величины с похожим названием, то это не означает, что величины имеют один и тот же физический смысл. Например, рейтинг депутата среди всего населения города vs. рейтинг только среди бабулек. И то и то рейтинг (что бы это ни было), эти числа (или что это там) могут даже сильно коррелировать между собой, но смысл у этих параметров всё равно разный, и эту разницу можно обнаружить.
- «Случайные» погрешности. Тут имеются в виду и некие систематические ошибки, о которых не известно экспериментатору/симулятору-теоретику, или действительно случайные ошибки в эксперименте/расчёте, которые невозможно контролировать и/или предсказывать. В принципе подобные вещи могут сами становиться предметом исследования различных интересных систематических эффектов.
Пример подобных исследованийНапример, есть история с непонятными сигналами на радиотелескопе
Спойлерубийца — микроволновка
которые оказались следствием слишком рано открываемой микроволновки
или же просто оценка полезнейшего соотношения S/N («сигнал/шум»). - И последней стандартной ошибкой является произрастание рук экспериментатора/расчётчика из тазовой кости, то бишь обычные человеческие ошибки. Тут исследовать ничего не надо, достаточно перепроверить работу или повторить произведённый эксперимент, чтобы обнаружить и устранить соответствующий косяк.
О последних двух типах ошибок ничего более конкретного сказать нельзя, но вот про первые две, да если взять конкретный метод исследования, наговорить можно очень много чего. Поэтому на них и сконцентрируемся. Основной упор при этом мы будем делать на структурных различиях молекул.
Ошибка #1. Различия молекулярных свойств в разных условиях
NaCl: когда ошибок не возникает
Вот почему-то никому не приходит в голову сказать, что у монокристалла поваренной соли (NaCl), представляющей из себя огромную молекулу из ионов Na+ и Cl–, и у двухатомной молекулы NaCl, получаемой испарением этого кристалла при бешеных температурах, одна и та же, скажем структура.
И даже если мы предположим, что хотя бы расстояния между хлором и натрием (rNaCl) и там и там одинаково, то эксперимент нас поставит на место:
- в двухатомной молекуле эта величина составляет rNaCl = 2.36 A,
- а в кристалле аналогичная величина будет rNaCl = 2.81 A.
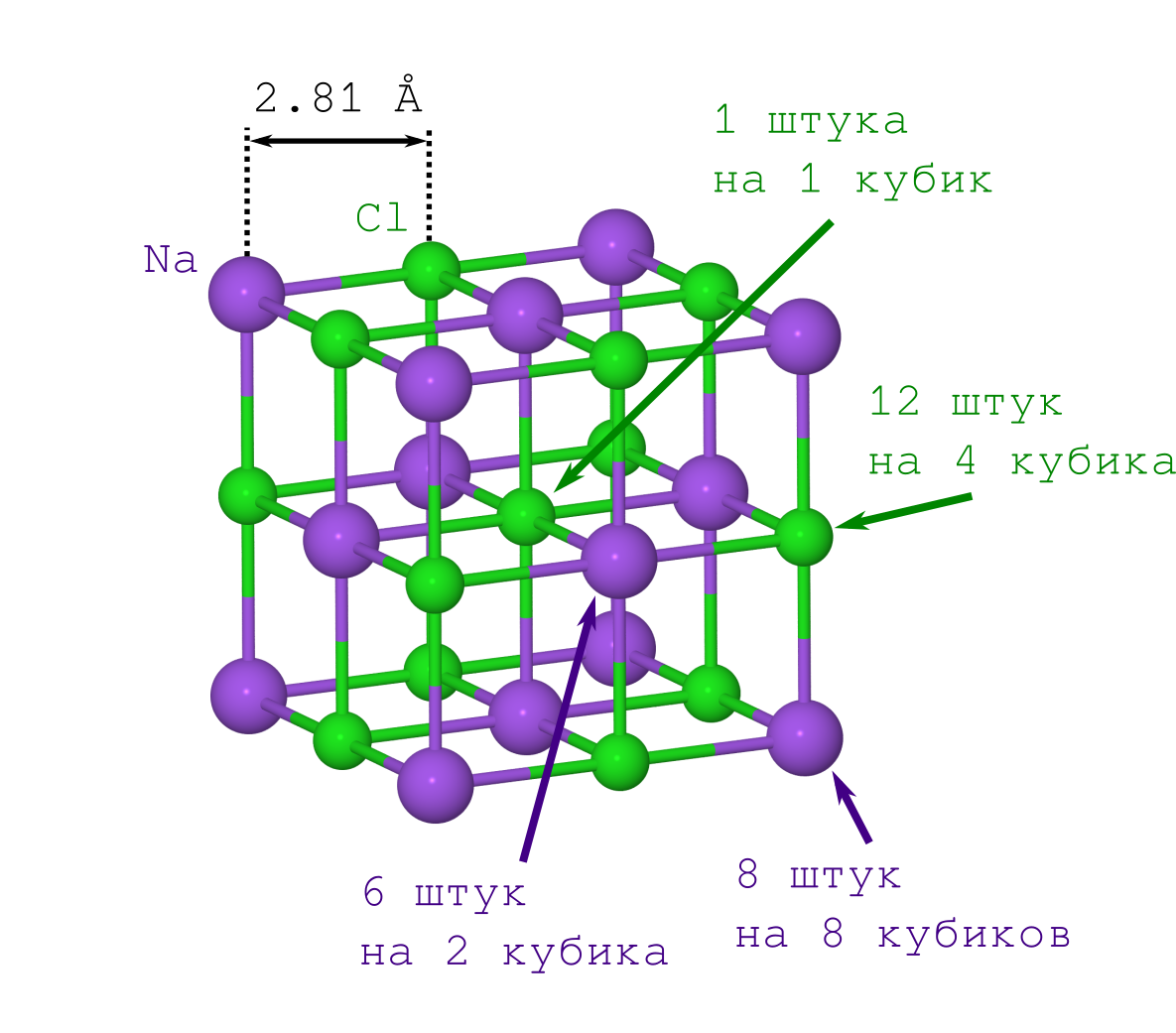
Но для получения расстояния между атомами в кристалле достаточно только плотности кристаллика поваренной соли ?=2.165 г/см3, которую можно как легко достать из Википедии, так и померить самому в домашних условиях.
Для вычисления расстояния нам понадобится:
- плотность кристалла NaCl (есть),
- знание расположения ионов этого кристалла.
Если бы делали это впервые (скажем, в начале XX века), то пришлось бы помучиться со вторым пунктом. Но современным людям это уже и так известно: решётка NaCl имеет форму кубика, в котором чередуются между собой ионы Na+ и Cl– (см. картинку выше). Размножая указанный фрагмент кристалла («копипастя» указанный кусок и прилаживая его к предыдущей итерации грань к грани), мы получим кристалл NaCl любого желаемого размера и любой желаемой (майнкрафтовой) формы.
А значит плотность этого кубика должна быть такой же, что и у всего кристалла. Учитывая, что плотность это (т.е. масса на объём), то получается, что зная массу и геометрическое выражение для объёма мы можем посчитать и расстояние между атомами.
Объём кубика очевиден: длина ребра составляет удвоенное расстояние Na--Cl (), а значит искомый объём равен .
С массой же не так всё просто. Большинство атомов у нас лежат на вершинах, рёбрах и гранях кубика, а значит одновременно принадлежат нескольким из этих кубиков. Это надо учесть при вычислениях.
Начнём учёт с ионов Na+. Их у нас всего 2 типа (см. рисунок решётки кристалла):
- те, что лежат в вершинах куба (их столько же, сколько и вершин куба, т.е. 8, и они одновременно лежат в 8 кубиках, поэтому надо будет делить это число на 8),
- те, что лежат на гранях (их 6 штук, и они одновременно принадлежат 2-м кубикам).
В итоге получаем, что в нашем кубе содержатся иона натрия.
Теперь о Cl–. Их тоже всего 2 типа (см. рисунок решётки кристалла):
- те, что лежат на рёбрах куба (их 12 штук, и они находятся в совместном владении у 4-х кубиков),
- тот Cl–, что в центре куба, он один и принадлежит только нашему кубику.
Поэтому нашем кубе содержатся иона хлора.
Состав кристалла, очевидно, соответствует химической формуле NaCl, но масса нашего кубика оказывается равна (не забываем, что массы атомов в таблице Менделеева даются в атомных единицах массы):
Теперь же из соотношения мы можем составить уравнение на длину :
,
которое легко решается:
.
От данных из рентгеновской кристаллографии 2.81 A (например из Abrahams, S.C.;Bernstein, J.L. Accuracy of an automatic diffractometer. Measurement of the sodium chloride structure factors // Acta Crystallographica (1965) 18, 926-932) мы промазали всего на 0.01 A, что достаточно круто.
Кто-то может подумать, что разница в 0.45 A несущественна, но это почти что боровский радиус (0.52 A), который равен наиболее вероятному расстоянию электрона, и по атомным меркам разница получается огромная.
В двухатомной молекуле 3s1 электрон натрия тоже хочет перепрыгнуть на атом хлора (ведь у них большая разность электроотрицательностей), но получающаяся разность зарядов тянет электрон обратно на натрий, в результате устанавливается «равновесие» между двумя резонансными структурами:
с ковалентной связью (слева) и ионной (справа), причём вторая более выражена.
Этот эффект можно измерить, поскольку реальный заряд на атомах будет не , как мы привыкли в школе, а нечто меньшее по модулю.
У молекулы NaCl в газовой фазе известны и расстояние между атомами (2.36 A), и дипольный момент , где и есть искомый модуль зарядов атомов (у натрия заряд будет , а у хлора ). Дебай для нас не очень удобен, поэтому перейдём к единицам «заряд электрона на ангстрем» при помощи множителя 0.21, т.е. , и в результате решения простейшего уравнения получим: . Вот эти «убежавшие» 0.2 заряда электрона и создают разницу между кристаллом NaCl и молекулой NaCl в газовой фазе.
Ферроцен
Стоит перейти от ионных кристаллов к молекулярным, в которых плотно упакованы молекулы, так сравнивать внезапно становится можно, причём без всяких оговорок.
Но о разнице не нужно забывать. И на эту тему имеется даже классический пример: молекула ферроцена.
Это простейшее сэндвичевое соединение. В нём нейтральный атом железа (как котлетка) зажат между двумя пятичленными ароматическими кольцами (булочками).
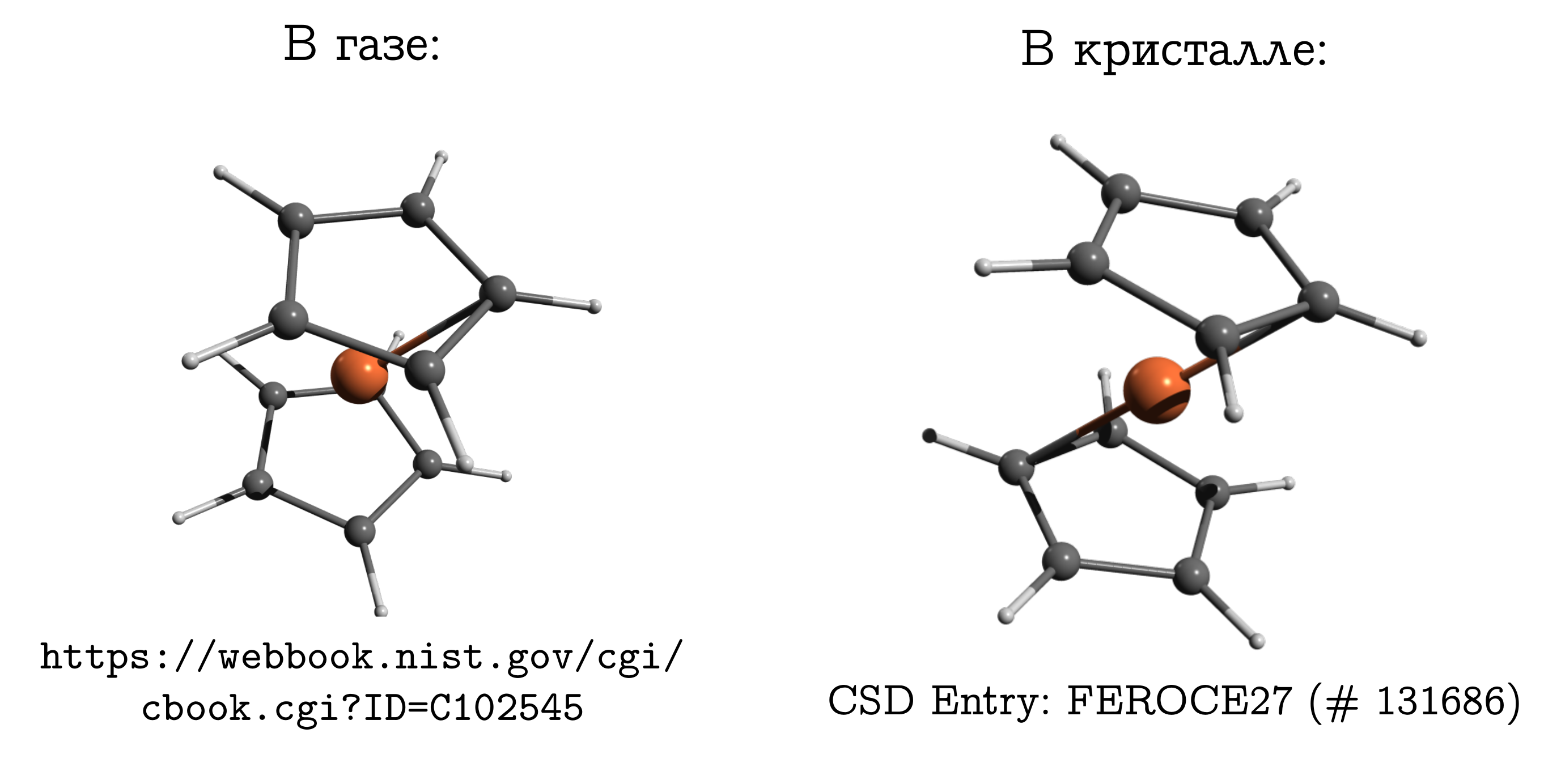
Эту молекулу можно достаточно легко испарить, и узнать, что наиболее стабильной структурой в газовой фазе является т.н. заслонённая конформация. В ней углероды и водороды верхнего и нижнего колец находятся друг напротив друга (см. картинку выше), поскольку в этом случае наиболее сильны дисперсионные взаимодействия между этими кусками молекулы, а дисперсионка — это всегда выгодно.
Если же мы возьмём кристалл ферроцена, то окажется, что там молекулы имеют другую стабильную конформацию (которую для углеводородов называют заторможенной), в которой водород и углерод одного кольца находятся над/под связью C--C другого. Между молекулами тоже есть дисперсионные взаимодействия, и подобная, вроде неудобная для молекулы, структура возникает из-за того, что молекулам проще уложиться близко друг другу только в неудобной для себя форме, а это личное неудобство компенсировать взаимодействием друг с другом.

Взято из www.chem.msu.su/rus/teaching/stereo
А здесь вся разница в расстоянии. Стандартной формой потенциала дисперсионных взаимодействий является потенциал Леннарда-Джонса (это, кстати, один, а не два мужика):
В нём первое слагаемое берётся из межатомного отталкивания, а второе — из межатомного притяжения, возникающего из флуктуаций электронной плотности. В целом, этот потенциал выглядит как-то так:
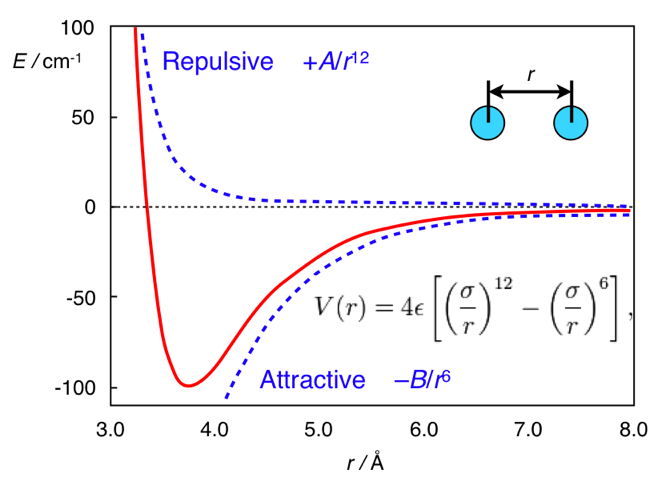
Потенциал Леннарда-Джонса. Взято из chemistry.stackexchange.com/questions/34214/physical-significance-of-double-well-potential-in-quantum-bonding
И в случае этана, атомы водорода находятся слишком близко друг к другу, поэтому они находятся (относительно её минимума) в левой части кривой, и для них характерно отталкивание. В случае же ферроцена, между кольцами есть прослойка нехилого размера (атом железа), из-за которого кольца оказываются достаточно далеко, чтобы не чувствовать межатомное отталкивание. И поэтому они находятся на правой (притягивающей) части потенциала.
Гистамин
В случае ферроцена мы увидели т.н. конформационные различия: молекула осталась одной и той же (т.е. никаких химических связей не порвалось и не образовалось), а её форма чуть-чуть изменилась.
Но отличия могут быть ещё более сильными, например, если в системе у нас возможны т.н. таутомерные превращения. Таутомеризация — это класс химических реакций, которые происходят так легко и быстро, что в итоге у нас могут одновременно существовать несколько изомеров одной молекулы, легко переходящие друг в друга. Эти изомеры называют таутомерами.
Стандартный пример подобного: кето-енольная таутомерия в кетонах:

Чаще всего, как и в этом примере, таутомерия связана с перескоком протона с одного тёпленького места на другое. И завязаны эти реакции на туннельный эффект, которому водород, как самый легкий из атомов, наиболее подвержен.
Подобные химические превращения характерны для многих биологических молекул, например для азотистых оснований, входящих в состав ДНК, или для сахаров.
Но вот при переходе от системы к системе часто меняются константы равновесия подобных реакций, поэтому в разных фазах мы можем наблюдать разный таутомерный состав.
Примером этого можно взять молекулу гистамина (см. рис. ниже).
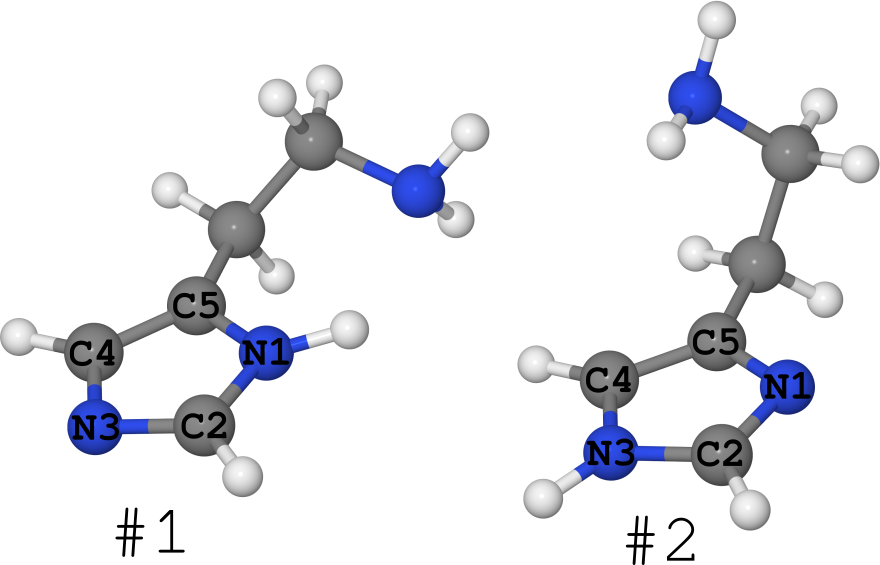
Она существует в виде 2-х таутомеров (про количество конформеров я вообще молчу, их очень много):
- #1, где водород сидит на азоте N1,
- #2, где водород сидит на азоте N3.
Так получилось, что для этой молекулы известна её структуры в разных фазах.
- В кристалле она находится полностью «замороженной» в форме #1. (см. статью DOI: 10.1021/ja00796a011 и структуру в кембриджском банке структур под именем «HISTAN» и/или номером 1176642)
- В водных растворах эта молекула существует в обеих формах, причём таутомера #2 заметно больше (DOI: 10.1021/ja027103x).
- В газе же гистамин существует в равной степени в форме #1 и в форме #2 (DOI: 10.1021/ja980560m).
Т.е. разные фазы содержат разное количество разных молекул, а значит они — разные системы.
Вывод об Ошибке #1

Главный вывод, который нужно сделать из приведённых примеров таков:
При сравнении расчётов в одной фазе с экспериментом в другой надо быть готовым к систематическим отличиям.
Это не означает, что не надо сравнивать: сравнивать надо, но просто надо критичнее относиться к обнаруженным различиям и/или совпадениям, и по возможности оценивать подобные эффекты.
Ошибка #2. «Зоопарк» молекулярных параметров.
Вторая ошибка вкратце описывается так: если параметры называются похоже, но не одинаково — это разные параметры.
Чтобы понять, в чём же заключается источник подобного несогласия теории и эксперимента, придётся поподробнее разобрать и те стандартные экспериментальные методы, которые используются для получения молекулярных параметров, и модели, которыми аналогичные величины обсчитывают чисто из теории.
И здесь мы вновь будем говорить только о структурах.
Как получают экспериментальные структуры молекул
Чтобы как-то ограничить себя, поговорим только о методах исследования структуры одиночных молекул, т.е. о газовой фазе.
Основных источников подобной информации у нас два:
- газовая электронография,
- микроволновая спектроскопия.
На каждом из этих методов остановимся чуть поподробнее.
Газовая электронография
Метод достаточно старый, своё начало он берет в 30-х годах XX века, когда немецкие учёные Марк и Вирль провели первые эксперименты по дифракции электронов на газе.
Мало кто знает, но этот метод исследования причастен к получению трёх нобелевских премий по химии.
- Питер Дебай в 1936-м году получил свою награду с формулировкой:
"[for his work on] molecular structure through his investigations on dipole moments and the diffraction of X-rays and electrons in gases"
Это единственное явное упоминание газовой электронографии в заслугах лауреата, и не спроста. Основное уравнение электронографии для интенсивности молекулярного рассеяния носит имя Дебая.
собственно, уравнение Дебая
Здесь обозначает интенсивность рассеяния электронов (или рентгеновских лучей, или других частиц) парой i-го и j-го атомов на расстоянии друг от друга, — координата рассеяния, связанная с углом рассеяния и длиной волны частиц , а — способность этой пары атомов к рассеянию дифрагирующих частиц.
И несмотря на то, что об этом замечательном физике помнят что угодно (модель ионных растворов, его модель для расчёта теплоёмкости кристаллов), но только не электронографию, главный научный приз он получил (в частности) за неё.
- Лайнус Полинг в 1954. Да, тот самый, получивший 2 личные нобелевские премии,
да ещё и посадивший весь мир на витамин C, Великий Полинг. В бытность своей работы в Калтехе он, в частности, занимался газовой электронографией (см., например, DOI: 10.1021/ja01873a047). И конечно, знание структурной химии свободных молекул помогло ему создать знаменитую теорию химической связи (но не будем тут принижать и его большой кристаллографический бэкграунд). - Одд Хассель, лауреат 1969 года. Свою 1/2 нобелевской премии он получил за открытие конформационного равновесия. Сделал он это на основе электронографического исследования циклогексана. Эта молекула существует в виде двух конформаций: кресло (chair) и ванная (в английской традиции — лодка, boat).

Отсюда: www.shapeways.com/product/N5FE298DS/cyclohexane-2-molecules-boat-and-chair-form
Эти варианты расположения атомов быстро переходят друг в дружку, но в то время об этом не знали, и считали, что должна реализовываться только одна из структур. Только вот электронографический сигнал никак не хотел описываться ни одной из этих структур, и только комбинация сигналов от обеих конформаций смогла объяснить наблюдающуюся дифракционную картину (об этом можно подробнее почитать в книге И. Харгиттаи «Откровенная наука. Беседы со знаменитыми химиками»).
Схема самого метода очень проста (см. картинку ниже).
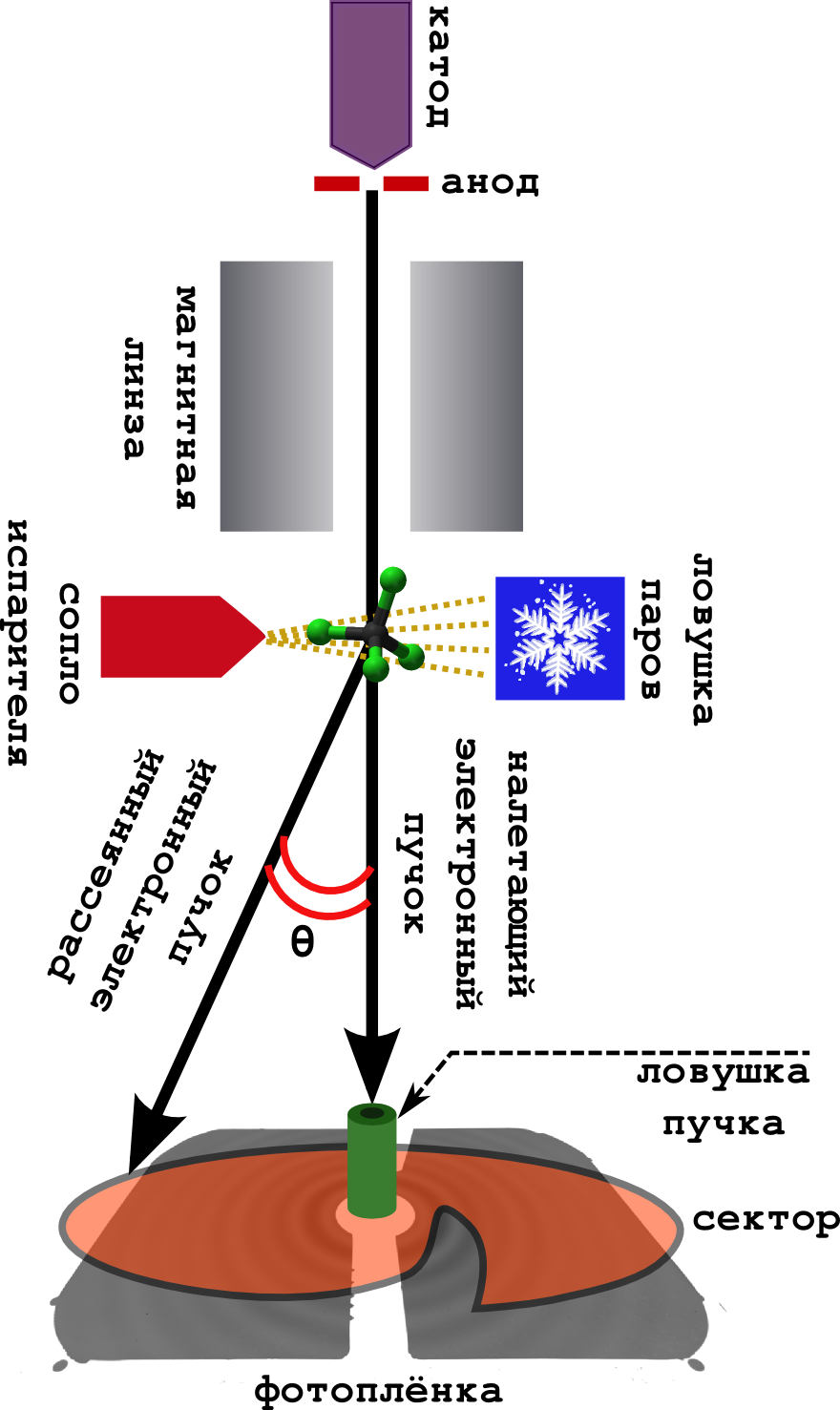
Дело происходит в вакууме.
- С катода непрерывно выбиваются быстрые электроны, которые ускоряются в поле анода до энергий 40-60 кэВ.
- Достаточно разбросанные (но быстрые) электроны фокусируются магнитной линзой, после чего они превращаются в узкий пучок.
- Перпендикулярно пучку установлена камера с веществом. Образец нагревается до кипения, и получившийся пар выходит на встречу электронному пучку.
- Электроны успешно рассеиваются на молекулах, и спокойно улетают дальше, где попадают на фотоплёнку.
- Обычно перед фотоплёкой ставят т.н. секторное устройство. Это очень быстро вращающаяся ширма необычной формы. Дело в том, что у электрона вероятность отклониться от изначального направления (на большой углол рассеяния ), очень быстро падает. Поэтому, чтобы сгладить это падение интенсивности, сектор равномерно затмевает центральную часть фотоплёнки, оставляя дальнюю часть открытой. В результате получается более равномерно засвеченная картина.
- Ловушка пучка ловит те электроны, что не рассеялись совсем (а их очень много).
- Ну, и чтобы молекулы не летали по всему прибору, пачкая его, их вымораживают на холодной ловушке, охлаждаемой жидким азотом.
В итоге получается та самая дифракционная картина из концентрических колец, описываемая уравнением Дебая (это сигнал). Из неё можно потом вытащить напрямую различные молекулярные параметры.
Но в РФ их две: Москве (на Химфаке МГУ), и в Ивановском Химико-Технологическом Университете.
Микроволновая спектроскопия
Этот метод исследования молекул известен больше, поэтому о нём я поговорю чуть более кратко, на примере наиболее современной модификации: спектрометра с Фурье преобразованием (хз как по-русски, короче Fourier-transformed microwave spectroscopy).
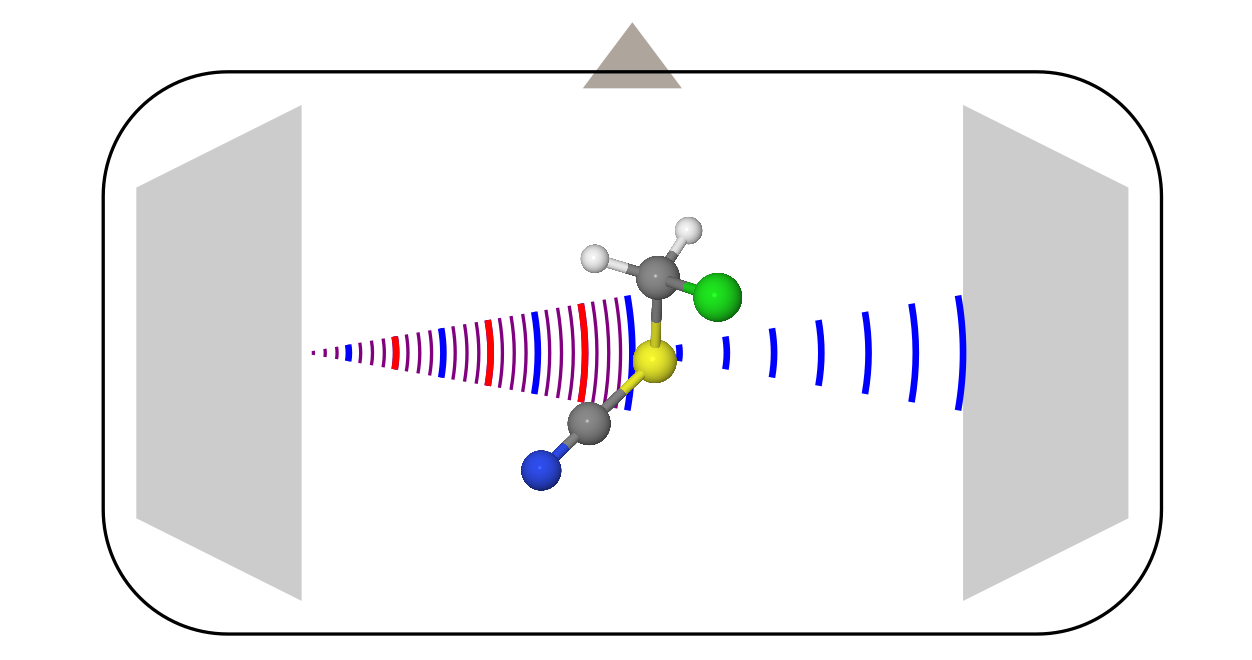
Конструкция тут уже более сложная, поскольку она требует кучу разной электроники (усилители, частотные модуляторы и т.д.). Всё это мы опустим, и поговорим только о том, что происходит внутри вакуумной камеры.
- Друг напротив друга стоят две рупорные антенны (наподобие той, которой открыли реликтовое изучение). Одна из них служит в качестве передатчика, а вторая является приёмником.
- Перпендикулярно этим антеннам стоит клапан, который запускает образец. Чаще всего он запускается в виде пара вместе с неким газом носителем (обычно это инертные газы) в режиме адиабатического расширения. В таких условиях молекулы быстро охлаждаются до температур около 0 К, что существенно упрощает спектр, делая его более податливым к интерпретации.
- Когда молекулы заполняют всю камеру, передающая антенна облучает их сигналом с линейной частотной модуляцией. В частотном представлении это соответствует сумме всех частот в некотором диапазоне.
- Некоторые молекулы поглощают это излучение на разных переданных частотах, и переходят в возбуждённое состояние. Но, через некоторое время они сваливаются обратно, начиная излучать то, что они нахапали при импульсе от передающей антенны. Это сваливание выглядит как убывающий осциллирующий сигнал (free induction decay). Вторая антенна его и регистрирует. Потом, после Фурье-преобразования записи этого сигнала во времени, получается обычный спектр от частоты.
В отличие от электронографии, которой было не важно, что за молекулы рассматривать, в микроволновой спектроскопии молекула должна обладать постоянным дипольным моментом (в редких случаях подходит и магнитно-дипольный момент, это характерно для радикалов, типа молекулы O2). Сигналом же тут является «интенсивность испускания vs. частота». Из этих спектров через некоторые модели извлекают вращательные постоянные, из которых уже потом достают структуру молекул.
Добро пожаловать в Зоопарк Молекулярных Параметров!
Теперь пришло время посмотреть на то, какие геометрические параметры мы можем вытащить из различных экспериментах. По-сути, каждый из типов величин обозначает что за модель была использована для подгонки экспериментального сигнала (чаще всего по методу наименьших квадратов). Большинство из приведённых параметров можно отыскать в обзоре Kuchitsu K., Cyvin S.J. // In: Molecular Structure and Vibrations / Cyvin S.J. (Ed.) — Amsterdam: Elsevier, 1972. — Ch.12. — P.183-211.
Начнём опять же с электронографии.
- -структура. Это просто набор средних значений межатомных расстояний при заданной температуре.
- эта величина аналогична , но она несколько более естественна для описания дифракционной картины.
- . Эта величина не имеет чёткого физического смысла и целиком завязана на интерпретационную модель. По-сути, это то, что наблюдается в кристаллографии.
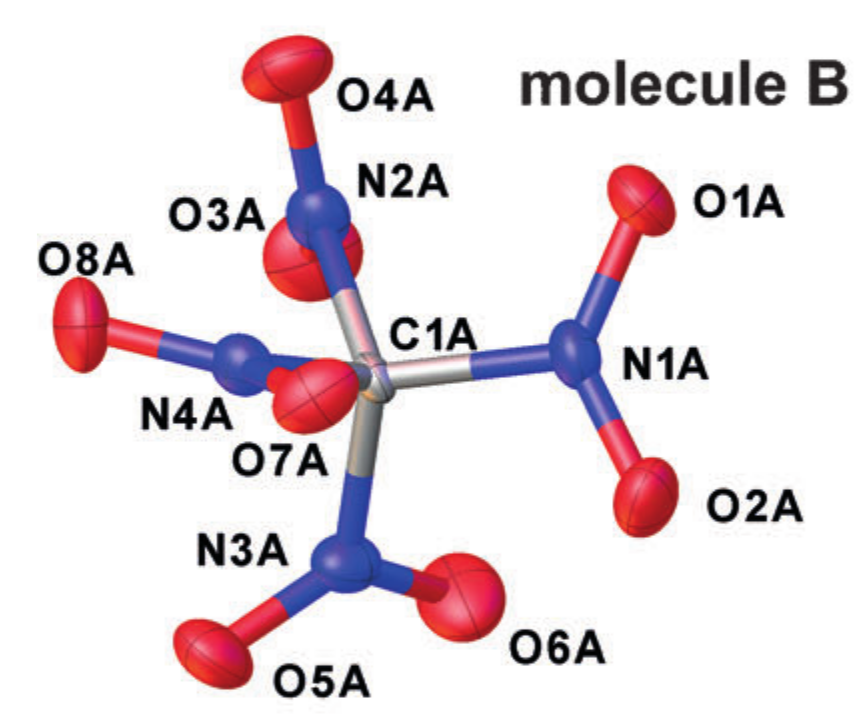
Пример кристаллографической -структуры (тетранитрометан в кристалле). Взято из DOI: 10.1002/anie.201704396
Каждый атом аппроксимируется эллипсоидом, описывающим его колебательное движение, и в качестве межатомных расстояний берутся расстояния между центрами полученных эллипсов. Но, подобное упрощение характера движения атомов соответствует введению приближения гармонического осциллятора для колебаний, а оно не всегда хорошо работает.
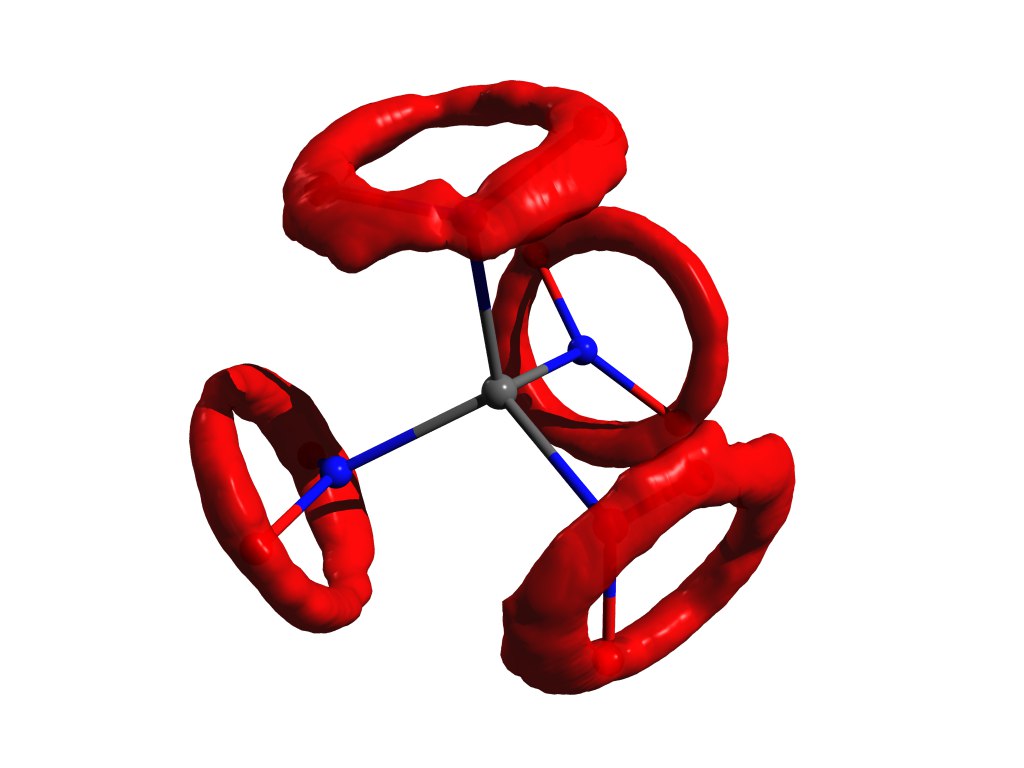
Пример распределения атомов, когда приближение гармонического осциллятора не работает. Тот же тетранитрометан, но в газе.
- . Эта
НЁХчудо-юдо рыба-кит в двух словах никак не описывается, к реальности имеет очень слабое отношение, но обладает замечательными свойствами: она должна быть геометрически согласованной (см. ниже) и её легко посчитать. За счёт этого она обрела свою большую популярность в электронографическом коммьюнити.
В микроволновой спектроскопии вариантов структур несколько меньше.
- Самой физически понятной является структура. По-сути, это усреднённая геометрия по некоторому колебательному состоянию молекулы (). Поскольку чаще всего работают с холодными молекулами, то наблюдают обычно одну конкретную структуру из этого класса: , т.е. геометрию молекулы в основном колебательном состоянии, когда атомы совершают нулевые колебания вокруг своего наиболее выгодного положения.
- Наиболее же популярной является -структура. Индекс «s» означает «substitution». Получают её так: полагают, что есть некоторые координаты атомов, которые фиксированы в пространстве, потом производят одноатомное изотопное замещение атома в молекуле, и по изменению вращательных постоянных устанавливают положение этого атома. Основным плюсом подобной технологии является простота. Минусом: нужно только монозамещение +не все позиции атомов можно так установить + не очень ясен физический смысл такой модели.
- Логичным развитием -структуры являются -структуры, которые получаются из подгонки по масс-взвешенному МНК. Для них тоже нужны изотопозамещённые молекулы, но тут уже подходят любые.
И это ещё далеко не все возможные типы структур…
Но
И вот встаёт вопрос: корректно ли сравнивать -структуру с какой-то из экспериментальных, смотря только на экспериментальные погрешности?
Ответ тут прост: нет, надо закладывать ещё дополнительную погрешность на возможные систематические отличия. И этому можно привести очень яркий пример: эффект Бастиансена-Морино (см. статьи DOI: 10.1107/S0365110060002557 и DOI: 10.1107/S0365110060002545).
Допустим, у нас имеется молекула типа CX2 (т.е. CO2, CS2 и т.д.). Как мы должны знать из курса школьной химии, эти молекулы линейную структуру (атомам углерода и два халькогена X лежат на одной прямой).
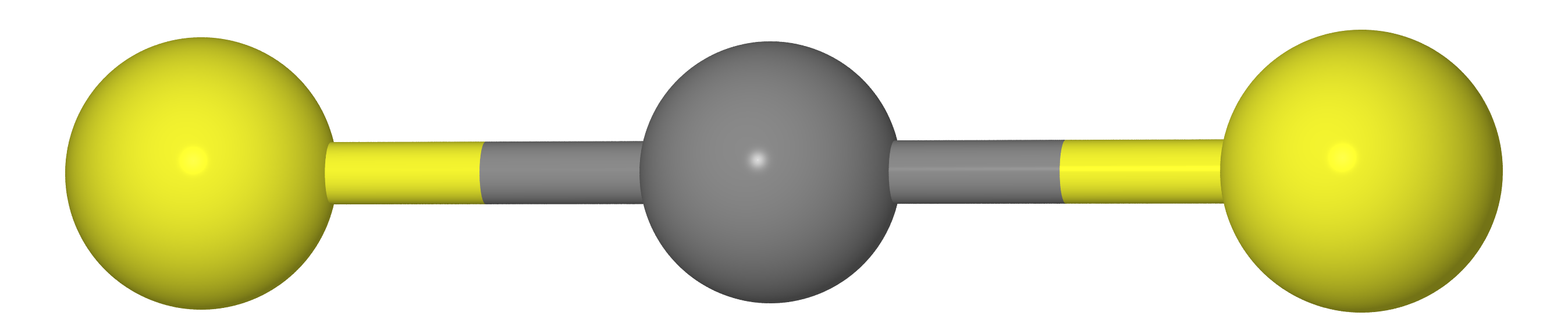
Значит, что расстояние между атомами X должно быть равно удвоенной длине связи C--X (т.е. ).

Как бы то ни было, если мы измерим расстояния между атомами C и X() и X-X () методом газовой электронографии, то получим, что , т.е. молекула получается как бы изогнутой. Причина кроется в том, что молекула совершает т.н. ножничные колебания, из-за которых атомы X оказываются существенно ближе друг к другу, чем в наиболее выгодном расположении (см. рис. ниже).

Откуда берётся эффект Бастансена-Морино. Картинка из статьи DOI: 10.1039/C6CP05849C.
Поэтому, если бы мы приравняли температурно-усреднённую -структуру к равновесной (), мы бы сделали неправильный вывод о том, что молекулы углекислого газа и сероуглерода — изогнутые.
Вот поэтому при сравнении разных типов геометрических параметров надо всегда быть очень аккуратным. Это относится как к сравнению между собой экспериментальных данных, так и к сравнению эксперимента и теории.
Косяки Стандартной Модели Молекулы
А теперь представим, что мы возжелали всем сердцем просимулировать результат некоторого эксперимента на основе нашей теоретической модели, дабы сравнить в честном бою симуляцию с реальностью.
И вот тут надо тоже быть аккуратным, т.к. разные модели молекул тоже имеют свои границы применимости. Разберём это на примере Стандартной Модели Молекулы.
Для начала бы надо понять, что же такое Стандартная Модель Молекулы. У БАКовских физиков своя Стандартная Модель, у астрономов — своя, вот и у физхимиков имеется своя базовая конструкция, от которой потом и пляшут. Но в отличие от физических моделей, то, что мы рассмотрим является набором приближений, позволяющих юзеру получить результат относительно автоматически и быстро.
Общая схема вводимых приближений выглядит как-то так:

В самом низу качества лежит наша стандартная модель. Кратко пройдёмся по всем этапам её получения.
- Во-первых, мы сразу забываем о существовании релятивизма, и остаёмся в рамках классической квантовой механики (ну и оксюморон). Что же тогда мы имеем? Есть молекула, состоящая из ядер и электронов, летящая в вакууме. Поскольку её никто не трогает, то мы можем ввести инерциальную систему отсчёта, отделив поступательное движение центра масс молекулы («Tr», от слова translation).
- После этого мы замечаем, что электроны у нас почти в 2000 раз легче ядер, поэтому под действием кулоновского притяжения к ядрам и кулоновского отталкивания друг от друга, они движутся куда быстрее ядер. В итоге электронам кажется, что ядра заморожены. Поэтому электрончики спокойно принимают наивыгоднейшую для себя конфигурацию, и стоит ядрам чуть-чуть подвинуться, так электроны уже перестроились, как им удобнее. Ядра же видят только некоторый усреднённый потенциал, создаваемый электронами и кулоновскими взаимодействиями всего со всем, а сами детали движения электронов не видят. Это разделение электронов и ядер носит название "приближение Борна-Оппенгеймера" (да, это тот самый Роберт Оппенгеймер, он не только бомбой известен).
- После отделения движений электронов («El») остаются колебания ядер в поле электронов, и вращение всей молекулы в целом. Разделить эти 2 движения в общем случае не получается, поэтому принимается приближение, что ядра далеко не уходят от своего самого выгодного положения (равновесной геометрии). В этом приближении можно сформулировать условия Эккарта, через которые удаётся разделить вращение молекулы и колебания. Вращающейся же системой оказывается молекула, замороженная в своей равновесной геометрии. И это приближение называется «жёсткий ротатор» («RR», от rigid rotor).
- Ну и на сладкое: общий вид потенциальной энергии для колебаний (создаваемой электронами) неизвестен. Поэтому, чтобы получить хоть какую-то аппроксимацию, эту энергию раскладывают в ряд Тейлора в районе точки минимума (т.е. в окрестности равновесной геометрии) до квадратичных членов:
Получившийся потенциал — это ни что иное, как энергия частицы на пружинке, поэтому решая уравнения движения мы получаем, что молекула — это набор пружинок, колеблющихся с разными частотами. Поэтому это приближение называется «гармонический осциллятор» («HO», harmonic oscillator).
Получившаяся модель носит название RR-HO(@BO). Приближение Борна-Оппенгеймера (BO) мы трогать не будем, а вот о жёстком ротаторе и гармоническом осцилляторе в рамках структурной химии поговорить придётся…
И основная проблема с этим приближением заключается в том, что молекула не жёсткая, и колебания у неё совершенно не гармонические. Соответственно, в реальности нам нужно приближение нежёсткого ротатора и ангармонического осциллятора. И ключевое слово тут «ангармонический», т.е. «не гармонический».
Поговорим о простейших молекулах: двухатомных. Их примеров много: HCl, HBr, HI, CO, O2, N2, и т.д. и т.п. Они выделяются из всех молекул тем, что у них имеется всего одно колебание: растяжение/сжатие межатомного расстояния.
И вот это расстояние между атомами мы и можем замерить в газовой электронографии (в варианте среднего по температуре, ) и во вращательной спектроскопии (усреднённое по, скажем, основному колебательному состоянию, т.е. ).
И теперь встаёт Главный Вопрос Вселенной Жизни и Вообще:
какой будет и в приближении гармонического осциллятора, и как это соотносится с равновесным расстоянием ?
Для ответа придётся посмотреть на поверхность потенциальной энергии для двухатомной молекулы:

- Если мы будем работать в приближении гармонического осциллятора, то наш потенциал окажется симметричным относительно положения равновесия: идти вправо или влево от него выглядит одинаково. Поэтому молекула во время колебаний будет ходить направо и налево с одинаковой частотой, в итоге средним положением у нас всегда будет оказываться положение равновесия.
- В реальности же, потенциал не очень похож на параболу, и для двухатомных молекул он имеет положение минимума и 2 асимптоты:
- вертикальную (при ), возникающую из-за кулоновского отталкивания ядер, поскольку электроны больше «не помещаются» между ними и не создают химическую связь,
- горизонтальную (при )
В итоге, если молекула сокращает свою длину связи относительно положения равновесия, она упирается в стенку, а если увеличивает, то падает на мягкий диван. А молекула не дура, она будет больше валяться на диване, чем биться о стенку. Поэтому колебание усреднённое по расстоянию будет больше, чем равновесное (), и это заметно: подобные смещения имеют порядок 0.01 A, что выше погрешностей измерения.
Поэтому, даже если мы захотим посчитать что-то более похожее на эксперимент, оставаясь в рамках Стандартной Модели Молекулы (RR-HO@BO), то мы ничего нового не получим, поэтому в сравнении будет участвовать та самая равновесная геометрия.
Вывод об Ошибке #2

Иллюстрация из статьи DOI: 10.1002/anie.201611308.
А вывод до жути прост и состоит из 2-х частей.
- При кооректном сравнении все величины должны иметь один и тот же смысл.
- Если же величины разные, то об этом не надо забывать.
Примеры ошибок в научных работах
«Индусские работы»
Собственно, главное место, где можно обнаружить подобное — это журналы низкого уровня. В них достаточно редко встречаются статьи с крутыми результатами, поэтому их облюбовали «ВедуЩие Исследователи» из стран второго и более Мира (страны БРИКС и их менее успешные последователи). Под журналами «низкого уровня» тут подразумеваются не те, что публикуют статьи типа «Корчеватель: алгоритм типичной унификации точек доступа и избыточности», а вполне себе уважаемые научные издания. В моей научной области наиболее известные «полупомойки» это:
- Journal of Molecular Structure от Elsiver-а (с IF=2.011),
- Journal of Molecular Modeling от Springer-а (с IF=1.507),
(есть и другие). Как видно, по формальным признакам, в российской науке они считаются весьма приличными изданиями. Но, туда поступает такой наплыв
В качестве иллюстрации, я взял свежий номер Journal of Molecular Structure и прошёлся по оглавлению, и вуаля:
S. Sathiya, M. Senthilkumar, C. Ramachandra Raja, Crystal growth, Hirshfeld surface analysis, DFT study and third order NLO studies of thiourea 4 dimethyl aminobenzaldehyde // J. Mol. Struct., V. 1180 (2019), PP. 81-88.
https://doi.org/10.1016/j.molstruc.2018.11.067
Общая структура подобных работ весьма незатейлива.
- «Варится» (но чаще тупо покупается на Сигме) некоторое вещество. В этой работе вещество таки сварили.
- Далее для этого вещества может производиться рентгенструктурный анализа (РСА), откуда получается структура молекулы в кристалле. Поскольку РСА — это стандартная процедура, а РСАшники (кроме тех, что белки и другие огромные структуры изучают) чаще всего работают в режиме конвейера, то ничего особенного тут нет. Поэтому, молекула обсчитывается какими-нибудь простейшими методами в крякнутой программе Gaussian, являющейся квинтэссенцией термина «квантовая химия для домохозяек». И структуры теоретической молекулы в газе сравнивают с экспериментальными данными РСА… ииииии тут получается комбо из 1й и 2й ошибки, т.к. исследователи зачастую не брезгуют что-нть вякнуть о качестве квантовой химии.
- Далее в аналогичном стиле добавляются ИК и рамановская спектроскопии, УФ/Vis, измерения.
- В стандартных молекулярных визуализаторах, типа (крякнутого) GaussView рисуются красивые картинки, но обязательно в плохом качестве.
- Не делается никаких выводов по существу: «мы много поэкспериментировали, много посчитали, привели таблицы и картинки мы молодцы, дайте нам конфетку.»
А вот из моих любимых: статья
M. Govindarajan, M. Karabacak, FT-IR, FT-Raman and UV spectral investigation; computed frequency estimation analysis and electronic structure calculations on 1-nitronaphthalene // Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, V. 85 (2012), PP. 251-260,
https://doi.org/10.1016/j.saa.2011.10.002.
В ней тупо снимался спектр в твёрдой фазе, а потом интерпретировался на базе средненьких расчётов в модели HO в газовой фазе. Но фишка в том, что они даже расчёты сделать нормально не смогли, о чём им вежливо намекнули в комментарии на статью.
Впрочем, термин «индусские работы» (как и термин "индусский код") относится далеко не только к работам, идущим с соответствующего загадочного субконтинента.
Если зайти на замечательный сайт Киберленинки, то
- "Водородные связи по верхнему и нижнему ободу молекулы в каликс[n]аренах (n = 4, 6) по данным ИК-спектроскопии и квантово-химических расчётов"
- "Подход к априорной оценке оптических спектров поглощения больших молекул",
- "Оценка адекватности квантово химических методов расчета азотсодержащих гетероциклических соединений данным РСА".
Особенно порадовала меня последняя, ведь «оценка адекватности» неадекватным сравнением структур в разных фазах (газ vs. кристалл) и с разными смыслами ( vs. ) — это и правда верх адекватности.
А бывает ли такое в хороших журналах?
Да, «ошибки» бывают и там.
Причём, в итоге всё закончилось вообще супер: публикацией офигенной статьи, где справедливость восторжествовала.
Речь пойдёт об одном из органических молекул с экстремально длинной одинарной С--С связью: об 1,1'-бисдиадамантане:

Так вот, экспериментальная длина центральной одинарной связи в 1,1'-бисдиадамантане A, почти на 0.08 A длиннее (это дофига)!
Это удлинение одинарной C--C связи возникает из-за того, что два алмазоподобных куска, которые эта связь держит, очень большие, поэтому они отталкиваются друг от друга. Но, как мы помним из примера ферроцена, у нас есть и притяжение за счёт дисперсионных (лондоновских) сил. И вот из-за нехилого размера этих половинок молекулы, возникает большое дисперсионное притяжение между кусками. Оно и не даёт разорваться этой центральной связи, поэтому данную молекулу можно (относительно легко) испарить, не сломав её по дороге.
Центральная однинарная связь C--C очень длинная, и поэтому было очень интересно сравнить как теоретические методы воспроизводят реальность. А реальность в первых статьях, вот они:
- Peter R. Schreiner et. al, Overcoming lability of extremely long alkane carbon–carbon bonds through dispersion forces //Nature (2011), V 477, PP. 308–311
- Andrey A. Fokin et. al, Stable Alkanes Containing Very Long Carbon–Carbon Bonds // JACS (2012), V 134, PP. 13641–13650,
была представлена только кристаллографическими данными. В результате сравнивая между собой сложно сопоставимые вещи, не делая должной поправки на различие параметров, они пришли в итоге к выводу, что длина центральной C--C связи в газе составляет 1.655 A, промахнувшись на 0.02 A. А это существенно больше погрешности эксперимента.
К счастью, в итоге они скооперировались со специалистами по этим вопросам, и в итоге получили правильный ответ (краткое популярное изложение этой работы можно найти и на N+1).
А нужно ли сравнение?

После всего, что я написал о корректности сравнений, может возникнуть резонный вопрос: а нужно ли тогда вообще сравнивать результаты расчётов и результаты экспериментов между собой?
Нужно! Ещё как нужно!
Есть известное высказывание (достоверного автора которого я не смог найти):
Nobody believes theoretical calculations, except the one who did them.
Everybody believes experimental results, except the one who obtained them.
В переводе на русский это звучит как: Никто не верит теоретическим расчётам, кроме того, кто сделал их, но все верят экспериментальным данным, кроме того, кто их получил.
А в науке нужно, чтобы верили все (ну или большинство), и эксперимент является единственным мерилом, поскольку соотносит то, что мы насчитали, с Реальностью.
На тему того, как же нужно поступать исследователю, или обывателю, читающему научные статьи и/или новости, есть замечательное эссе (в открытом доступе) во втором по значимости химическом журнале: Mata R., Suhm M. // Angew. Chem. Int. Ed., 56 (2017), DOI: 10.1002/anie.201611308
(кстати, ссылку на него я уже приводил, поскольку одна картинка, за авторством Рикардо Мата, как раз из этой статьи).
В выводах этого эссе приводятся рекомендации теоретикам-симуляторам и экспериментаторам. Их я и приведу тут (в переводе и небольшой переработке) в качестве завершающего слова к данному посту.
- Теоретик должен:
- приводить не только успешные свои методы и попытки, но также описывать фейлы методов (особенно, если эти методы популярны),
- хорошо и полно описывать свою методологию,
- где возможно приводить оценки (или описание) погрешностей, и важные принятые приближения и упрощения.
- Экспериментаторы, в свою очередь должны:
- совать в нос теоретикам свои экспериментальные данные, которые они бы могли использовать в качестве бенчмарков (эталонов),
- показывать научному сообществу непонятные экспериментальные данные, где теория (или дополнительные эксперименты) помогли бы с объяснением,
- выступать на
чужой территориитеоретических конференциях со своими данными,
личная историяКстати, с вторым автором этой статьи, который является экспериментатором, я как раз и познакомился на теоретическом симпозиуме.
- вытаскивать из своих экспериментальных данных вещи, максимально доступные для сравнения с теорией,
Всех благ и корректных сравнений! И помните: не ошибается только тот, кто ничего не делает.
P.S.
В качестве послесловия хотелось бы привести небольшой списочек баз данных, где можно откопать разные экспериментальные данные для молекул.
Структурные банки данных
Кристаллографические банки
Проще всего определить структуру молекулы в кристалле, т.к. РСА — это рутинная процедура. Поэтому если не знаете, как выглядит молекула, вперёд в кристаллографические банки данных (места, где собраны почти все структуры веществ, когда-либо запиханных в гониометр, и освещённых пучком коротковолновых частиц). Поскольку таких банков очень много, приведу только самые известные (более полный список можно надыбать в Вики).
- Inorganic Crystal Structure Database (ICSD). Это не совсем про молекулы, там в основном можно найти структуры разных солей, металлов, керамики и т.д. Поддерживается эта база Карлсруэвским Технологическим Универом, поэтому доступ к ней платный и недешёвый. Но если что, сайт её.
- Cambridge Structural Database (CSD). Пожалуй, самая большая кристаллографическая база в Мире. Почти миллион структур! В основном это органические и элементорганические молекулярные кристаллы. И эта база бесплатная! За доп. плату, конечно, можно получить клёвый опционал, типа поиска по нарисованной структуре молекулы, но это уже излишки. Сайт.
- Crystallography Open Database (COD). Ну тоже какая-то база данных. Что там есть, я особо не знаю, но она, по крайней мере, существует. Сайт.
- Ну и для тех, кого интересует биология, есть Protein Data Bank (PDB). Открытая база данных, откуда можно скачать структуры огромных и страшных белков (но в кристаллах). Сайт.
Структуры молекул в газе
Тут дела обстоят несколько хуже, т.к. эксперименты по изучению структуры свободных молекул — существенно более сложны, как для проведения (как минимум нужен высокий вакуум), так и для интерпретации.
Поэтому и баз данных тут существенно меньше.
- Самая большая база данных для свободных молекул — это MOlecular GAsphase DOCumentation (или MOGADOC). Базируется она в Ульмском Университете, и является весьма недешёвым вложением. Но, если что, сайт вот.
- Если же хочется узнать 100% экспериментальные равновесные структуры молекул, то это в NIST-овскую Computational Chemistry Comparison and Benchmark DataBase (CCCBDB). Почти все чисто экспериментальные -структуры можно найти там, но и других ништяков там хватает. Сайт.
- Последний оплот — это слабо развивающийся проект MolWiki. Но там особо много структур не найти, только те, что были получены в Университете Билефельда, МГУ и ИГХТУ за последнее время (да и то не все). Сайт.
Где найти спектральные характеристики молекул?

Взято из xkcd.com
Тут баз данных существенно больше, поскольку снять спектр без интерпретации — это существенно более простая задача, чем получение структуры (не нужно строить модели и доказывать, что они верные). И к тому же, спектры имеют огромное прикладное значение: по ним можно определить состав образцов, будь то водичка из соседней речки, или сигнал из молекулярного облака (или даже из атмосферы экзопланеты, см. иллюстрацию выше).
Да, кстати, все ссылки в этом разделе будут на бесплатные базы данных.
- NIST Chemistry WebBook. Пожалуй, лучшая книга в мире. В этой базе данных можно найти ИК, UV/Vis, масс-спектры для кучи молекул. А также, термохимические параметры и даже иногда кинетические! В общем, если химик не знает о существовании этого сайта, то он не химик. Сайт.
- High-resolution transmission molecular absorption database (HITRAN). Проект Гарвардско-Смитсоновского центра астрофизики, из чего очевидно, что спектры, приведённые там, используются для идентификации молекул в межзвёздном и околозвёздном пространствах (например, так). Сайт.
- Аналогичным предыдущему, является проект The Cologne Database for Molecular Spectroscopy, от астрофизической части Кёльнского универа. Сайт.
- Ну и в качестве последнего примера приведу проект ExoMol. Это не совсем чистые экспериментальные спектры, но это отличный пример взаимодействия теории и эксперимента: на основе высокоточных экспериментальных данных и очень высокоуровневых расчётов предсказываются спектры простейших молекул при разных (в т.ч. и экстремальных) условиях. Основной упор тут идёт на биомаркеры, поэтому, когда астрономы разглядят спектры экзопланет, они смогут легко идентифицировать в них уже известные нам молекулы. Сайт.
P.P.S.
Если есть ошибки/что-то осталось непонятным, пишите в комментах — исправлю/постараюсь объяснить получше.
Комментарии (61)

Victor_koly
14.12.2018 20:26С частотой радио- и микроволн (и даже длинноволнового инфракрасного излучения, ИК=IR) вращаются разные молекулы в газовой фазе
Возможно, что Вы и правы в такой формулировке (или это совпадение), но хотелось бы дать уточнение.
По аналогии со спектром энергий квантового осциллятора есть спектр вращательного движения.
Так вот, вращательные уровни характеризуются не «вращением на частоте микроволн», а разницей уровней вращательного движения такой, что
cB[(J+1)(J+2)-J(J+1)] = 2cB(J+1) ~ omega (частота кванта ЭМ излучения, например — 300 ГГц).madschumacher Автор
14.12.2018 21:01Да, Victor_koly, Вы правы, строгая формулировка именно такая.
Впрочем, процитированные слова я писал не с бухты барахты. И предпосылка у них следующая.
Как мы знаем, классическую вращательную энергию можно представить как , где L — момент импульса, а I — момент инерции. В выражении же через угловую скорость она записывается как
, где L — момент импульса, а I — момент инерции. В выражении же через угловую скорость она записывается как
 ,
,
где ? — угловая частота.
В квантовом случае (для линейных молекул) вращательная энергия имеет вид
") ,
,
где J — вращательное квантовое число. Частота перехода, как Вы правильно отметили, будет

Если же мы попытаемся оценить по классической формуле угловую скорость вращения молекулы (а мы имеем право так делать), то получим
} \underset{J \rightarrow \infty}{\rightarrow} \frac{\hbar J}{I}") .
.
Иными словами, при достаточно больших J угловая скорость вращения (а значит и энергия вращения) и частота возбуждающего излучения совпадают (классическая интерпретация говорит о том, что ЭМ волна должна войти в резонанс с вращением, чтобы провзаимодействовать), а энергия фотона и полная энергия вращения отличаются только на множитель 1/2. ;)
carpaccio
14.12.2018 21:40" И завязаны эти реакции на туннельный эффект, которому водород, как самый легкий из атомов, наиболее подвержен."
Откуда дровишки про то, что таутомерия «завязана» на тоннельный эффект? Это, кхм, крайне необщепринятое мнение. В отличие от множества примеров доказанных межмолекулярных механизмов.madschumacher Автор
14.12.2018 22:02Откуда дровишки про то, что таутомерия «завязана» на тоннельный эффект?
Из профессиональных знаний. В реакциях переноса протона (при температурах порядка комнатной и ниже) чаще всего присутствует или туннельный эффект (при повышении температуры его доля падает), или вообще происходит надбарьерный перенос (что, впрочем, ни что иное как «злой брат-близнец» туннельного эффекта). Это тупо из-за того, что термическая длина волны де Бройля у протона очень большая (порядка 1 A при T ~ 300K).
Это, кхм, крайне необщепринятое мнение.
Среди органических химиков может быть, почему бы и нет.
В отличие от множества примеров доказанных межмолекулярных механизмов.
1. стандартные картинки переноса с интермедиатами и промежуточными веществами никак не отменяют квантовых эффектов, необходимых при численных расчетах скоростей реакции,
2. если говорить о классических (то бишь известных испокон веков) механизмах из учебников по органике, то их доказательная база часто не соответствует современным критериям, и при рассмотрении их с современных позиций картинка может существенно меняться.
aslepov78
14.12.2018 21:44задам может глупые вопросы… Вот к примеру смоделировать механическую систему я могу с помощью физ движков вроде Bullet. Я понимаю какие там уравнения, и самое главное понимаю что движок просто ищет состояние системы в каждый след момент времени. Ок, квантовая механика вроде тоже имеет уравнения второго порядка (ур Шрёдингера). И вроде бы тоже можно численно чтото симулировать. Но вот когда встает вопрос а что именно можно расчитать — у меня ступор. Вопрос — откуда столько специфичных терминов в вашей классификации моделей? Они возникают из желания построить аналитическую модель? А если численно, уравнения Шрёдингера достаточно?
madschumacher Автор
14.12.2018 22:28И вроде бы тоже можно численно чтото симулировать.
Так об этом и речь! Только численно и получается обычно…
Вопрос — откуда столько специфичных терминов в вашей классификации моделей?
Хочется дешево и сердито. Какая-нибудь условная молекула воды состоит из 3-х ядер (3*3-6 = 3 степени свободы) и ещё 10 электронов (+ ещё 30 степеней свободы). Причём, поскольку это квант.мех, тут нет возможности задать начальные условия, а потом отслеживать поведение системы. Поэтому и приходится извращаться, чтобы получить результат за разумное время.
А если численно, уравнения Шрёдингера достаточно?
В лоб — очень дорого (подъёмно только для очень маленьких систем, а хочется симулировать химические реакторы, да живые клетки). И так, даже для этих приближений зачастую приходится гонять суперкомпьютеры неделями…aslepov78
14.12.2018 22:37>Причём, поскольку это квант.мех, тут нет возможности задать начальные условия
Секунду. Если мы говорим об уравнение Шреденгера, а это дифф ур 2го порядка, то там другого выхода и нет. Как можно вычислять след состояние не имея начального? В случае квант мех начальное состояние задается волновой функцией в нач момент времени.
>В лоб — очень дорого
Ну это понятно. Прежде чем искать что то оптимальное хорошо бы понять процесс считая что имеем неограниченные выч взможности. Даже с такими допущениями то масса вопросов. Может ссылочку подкинете где бы был вот такой ход, от примитивного к сложному. Еще вопрос — а для квант мех есть чтото наподобие физ движков вроде Bullet?madschumacher Автор
15.12.2018 00:26По-сути, geisha и mobi всё уже рассказали, так что ограничусь парой примерчиков.
Еще вопрос — а для квант мех есть чтото наподобие физ движков вроде Bullet?
Конкретно аналогичных нет, но есть свои, заточенные под специфичные задачи. Про всю квантовую химию не скажу, но для электронной задачи (грубо: поиска того, как электроны сидят по уровням энергии в молекуле) есть куча разных пакетов. Наиболее известным является Gaussian, но он жутко дорогой, да и скоростью не блещет. Из бесплатных могу выделить Orca, CP2K. Есть и хорошие (и тем более бесплатный) отечественные продукты от наших сумрачных гениев: Firefly от А.Грановского и Priroda от Д.Лайкова. В принципе, список подобных софтов доступен в Вики.
Для других же задач, более специфичных, приходится часто писать свой софт, причём распространена ситуация: 1 задача — 1 код. Уж больно это обширная область исследования, волшебной кнопки «всё сделать хорошо» тут ещё не изобрели.
Мне понять хочется откуда сложность.
Имхо, хорошей иллюстрацией служит формализм квантовой механики отSt.Фейнмана (интегралы по траекториям)
Чтобы вычислить в рамках классической механики, куда при заданных начальных условиях перейдёт система, надо просто проинтегрировать систему ОДУ (уравнения Ньютона, Лагранжа-Эйлера, Гамильтона — не важно).
В квантах же (в формализме интегралов по траекториям), чтобы вычислить какой «процент частиц» (не будем сейчас в этом вдаваться) перейдёт из точки A за время t в точку B, необходимо просуммировать все возможные и невозможные траектории системы между этими 2мя точками, причём с определёнными комплексными весами. И это только для 2-х отдельно взятых точек.
По-моему аналогия вполне понятна. :)aslepov78
15.12.2018 11:26Да, аналогия с траекториями понятна. Непонятно другое, где то место перехода от дифф уравнения 2го порядка, в частных производных, к матрицам и переходам. Конечно и помимо квантов полно задач где есть состояние, вероятности переходов между ними и надо найти состояние через время T. Как задача, где задано поле волновой функции, ее начальное состояние, — вдруг превратилась в задачу перебора всех вариантов переходов? Да да, знаю что с этого все и начиналось, про дискретные уровни электрона, переходы… потом появляется ур Шредингера, потом его решают, получают собственные функции и корни, и они типа и есть эта дискретная структура… Но вот этот момент непонятен — что мне мешает решать задачу как ур. в частных производных и искать эту функцию как тепловое или эл магн поле? В тех же полях нет этого комбинаторного взрыва переходов, откуда они при решении ур Шредингера?
madschumacher Автор
15.12.2018 13:53Но вот этот момент непонятен — что мне мешает решать задачу как ур. в частных производных и искать эту функцию как тепловое или эл магн поле? В тех же полях нет этого комбинаторного взрыва переходов, откуда они при решении ур Шредингера?
Временное уравнение Шрёдингера — это уравнение теплопроводности с комплексным коэффициентом теплопроводности.
Насколько часто Вам нужно решать уравнения Максвелла или уравнение теплопроводности, скажем, в 30-ти мерном пространстве? Или всё же за рамки 3D (время не считаем) в реальных случаях выход не требуется?
Сейчас решение «в лоб» можно потянуть, ну для 10-15-мерных случаев. А дальше никак.aslepov78
15.12.2018 17:10уравнение Шрёдингера — это уравнение теплопроводности с комплексным коэффициентом теплопроводности.
А что из этого следует? К комплексным уравнениям часто переходят исключая временную составляющую. В электродинамике например так делают сплошь и рядом. Да и в самом ур Шредингера так делают исключая переменную t считая поля гармоническими. Дело ведь не в комплексности. или не понял ваш ответ.
Насколько часто Вам нужно решать уравнения Максвелла или уравнение теплопроводности, скажем, в 30-ти мерном пространстве
Даже 1мерное уже не часто. Значит вы полагаете дело только в размерности задачи? Но размерность я могу и в классивеской механике создать, или в той же электродинамике. Размерность ведь параметрическая. Систему с многими параметрами запросто можно придумать. Но я же не про это спрашиваю. А про то откуда в принципе возникают вероятности переходов, матрица этих вероятностей и как следствие задача становится экспоненциальной. Т.е. в основе нормальное уравнение, 2го порядка, в частных производных. Но проблемы почему то совершенно другие чем в электродинамике или механике.madschumacher Автор
15.12.2018 18:28Дело ведь не в комплексности. или не понял ваш ответ.
Комплексность тут ни при чём, это я для строгости написал. А суть утверждения была в том, что для решения уравнения Шрёдингера применяются методы решения, что и для уравнений теплопроводности. В частности, стационарное уравнение Шрёдингера — это просто результат применения метода Фурье.
Значит вы полагаете дело только в размерности задачи?
Да. Только полагать тут и не требуется ничего, это факт.
Но размерность я могу и в классивеской механике создать,
Но Вам уже ответили, что решать уравнения классмеха (ОДУ) существенно проще, чем решать уравнения квант.меха (УрЧП). Проинтегрировать уравнения движения для десятков (или даже сотен) тысяч классических частиц — вполне посильная задача.
или в той же электродинамике.
Это как?! Уравнения Максвелла — это уравнение на 2 векторные функции в 3-х мерном пространстве. Откуда Вы ещё дополнительные размерности возьмёте (только если Вы не струнный теоретик)?
Размерность ведь параметрическая.
Ничего подобного. Размерность определяется изучаемой системой. Грубо говоря, в механике размерность — это число частиц умноженное на 6 (3 координаты и 3 импульса). В квантовой механике — число частиц, умноженное на 3 (координаты или импульсы). Но в первом случае нужно всего-лишь интегрировать диффуры, а во втором — уравнения в частных производных. И второе существенно гемморойнее.
А про то откуда в принципе возникают вероятности переходов, матрица этих вероятностей
Это не настолько просто объяснить в 2-х словах.
Т.е. в основе нормальное уравнение, 2го порядка, в частных производных. Но проблемы почему то совершенно другие чем в электродинамике или механике.
Ну потому что размерность сопоставима с тем, что нужно в механике, а уравнения настолько же сложные, как в электродинамике. К-к-к-к-комбо.
mobi
16.12.2018 12:17Значит вы полагаете дело только в размерности задачи? Но размерность я могу и в классивеской механике создать, или в той же электродинамике.
Да, но в классике всего 4 аргумента (x, y, z, t) и более высоких размерностей как правило не бывает, причем в механике все величины вообще зависят только от времени t. Уравнение Шредингера для 2 частиц (7 аргументов: x1, y1, z1, x2, y2, z2, t) в принципе тоже решают. Но как только число аргументов искомой функции переваливает за, например, 20-30, у вас уже просто нет памяти хранить все узлы сетки (например для системы из 10 электронов у вас 30 аргументов и если рассмотреть 10 разбиений на каждый, тогда нужно хранить 1030 узлов, сами посчитайте, сколько это потребует памяти для типа complex float).aslepov78
16.12.2018 12:24Спасибо. Дошло. Я упустил тот факт что частица становится волновой функцией а функция должна быть задана в каждой точке пространства.
Victor_koly
16.12.2018 16:15Задана в каждой. Ещё в идеале после каждого расчета проверять, что сумма квадратов модулей ВФ по ячейкам пространства, умноженных на объем ячейки дает 1. Либо же говорить, что у нас вышло чуть меньше 1, то есть мы посчитали вероятность того, что частицы покинут объем задачи. Скажем в том же угарном газе 1 электрон улетит на 100 ангстрем от ЦМ молекулы, а мы в виде объема взяли только цилиндр радиусом 50 А и высотой 50.
geisha
14.12.2018 22:41(автор вверху пишет то же самое с теми же примерами)
Вопрос — откуда столько специфичных терминов в вашей классификации моделей?
Из-за практической сложности симуляции на компьютере. Даже, к примеру, модель воды симулировать сложно. Одна молекула воды для уравнения Шрёдингера — непосильная задача, поэтому вы делаете каскад приближений. Каждое такое приближение — отдельный термин в модели. В результате получается классификация моделей воды, каждая — со своей областью применимости, с сильными и слабыми сторонами.
Они возникают из желания построить аналитическую модель?
Да, из желания построить достаточно простую модель, которую можно запихнуть в компьютер.
А если численно, уравнения Шрёдингера достаточно?
Нет. В примере с водой уравнение Шрёдингера вообще стараются не использовать. Это невообразимо сложно для компьютера. Вообще, было бы довольно глупо, если бы сложные квантовые модели симулировались с помощью заведомо более простых классических компьютеров без каких-либо приближений, не так ли?aslepov78
14.12.2018 22:51Что это сложно и не возможно я итак слышу. Мне понять хочется откуда сложность. Вот выше автор говорит о большом количестве степеней свободы. Но в классической механике они тоже никуда не делись. Тем не менее система из шариков на пружинках запросто моделируется. И в квантах и в классике уравнения 2го порядка, одинаково необходимо начальное состояние… Верю что там трудности принципиальные, иначе бы давно все симулировали молекулы. Но вот откуда они происходят, начиная с уравнения Шредингера (уж простите что с универа запомнил как фундамент квантов) — не понятно.
mobi
14.12.2018 23:21+1В классической механике обыкновенные дифференциальные уравнения, а в квантовой — в частных производных. Решить систему из 30 ОДУ в ? раз проще, чем одно уравнение в частных производных от 30 переменных.
aslepov78
15.12.2018 11:17В частных производных усложняет но ни это подозреваю проблема. Тепловые поля, электромагнитные — пример решаемых задач.
Victor_koly
15.12.2018 15:26Классическая механика, например — уравнение Эйлера-Лагранжа.
Берем например очень простую молекулу — CO.
Имеем 2 ядра и 14 электронов.
Если совсем без приближений — нам необходимо записать 16 уравнений, в которых потенциальная энергия будет участвовать в 16*15/2 = 120 членах. Получим уравнения типа:
m1*dr1/dt = dU/dr1,
где r1 — вектор (x1,x2,x3) и под производной по вектору имеется в виду градиент. Да, я ещё забыл учесть возникновение магнитного поля от движения зарядов — может членов в уравнении ещё больше станет; ещё забыл магнитные моменты ядер, но наверное для расчета молекул можно пренебречь.
Но есть хорошая штука — адиабатическое приближение. Точно смысла не помню, но могу предположить такой порядок действий:
1. Фиксируем положения ядер — в моем примере они весят не менее 12 а.е.м., что много больше массы 1 электрона.
2. Рассчитываем эволюцию электронов в потенциале 2 неподвижных ядер — задача «14 тел + потенциал».
3. Рассчитываем, как все же будут смещаться ядра под действием потенциала электронов из пункта 2.
4. Пробуем в пункте 2 учесть изменение положения ядер (и далее по циклу).
Считаем до тех пор, пока реальные изменения каждой новой итерации не превысят погрешности от того, что мы не посчитали релятивистскую поправку к движению четырех 1s-электронов вокруг ядер.Hilbert
15.12.2018 15:51Но есть хорошая штука — адиабатическое приближение. Точно смысла не помню
Ну, вы его в принципе описали. Идея в том, что раз электроны и ядра «живут» на совершенно разных временных масштабах, можно считать, что ядра живут в поле, усредненном по всем положениям электронов, а электроны живут в поле неподвижных ядер.Victor_koly
15.12.2018 16:13В таком смысле, если мы применяем ад. приближение к квантовой механике (точно помню, что учили в том курсе), можно потом понятие «поле электронов» записать в смысле том, что есть потенциал, вызванный распределением заряда
ro = e *|psy®|^2, а потом — записать действие на каждое из ядер потенциала
phy (r') = int{ro®/|r-r'|dr}.
Но кажется очевидным, что подобный гамильтониан с кин. энергией T
H = T1 + T2 +{потенциал отталкивания 2 ядер} + e*{phy (r1) + phy (r2)}
будет не очень легко считать.madschumacher Автор
15.12.2018 16:29Если что, в тексте есть про приближение БО = адиабатическое приближение.
После этого мы замечаем, что электроны у нас почти в 2000 раз легче ядер, поэтому под действием кулоновского притяжения к ядрам и кулоновского отталкивания друг от друга, они движутся куда быстрее ядер. В итоге электронам кажется, что ядра заморожены. Поэтому электрончики спокойно принимают наивыгоднейшую для себя конфигурацию, и стоит ядрам чуть-чуть подвинуться, так электроны уже перестроились, как им удобнее. Ядра же видят только некоторый усреднённый потенциал, создаваемый электронами и кулоновскими взаимодействиями всего со всем, а сами детали движения электронов не видят. Это разделение электронов и ядер носит название «приближение Борна-Оппенгеймера» (да, это тот самый Роберт Оппенгеймер, он не только бомбой известен).
Конечно, в некоторых источниках адиабатическим приближением называют чуть модифицированную версию этого явления, но сути это не меняет.
Подробнее про БО можно почитать в Давыдовском учебнике по квантмеху, или, например, тут.
aslepov78
15.12.2018 17:01я не очень понимаю зачем вы пересказываете решение в классической механике. Там мне как раз все понятно: есть система частиц, есть поля как посредник взаимодействия, есть силы. Все это и без приближений хорошо симулируется. Мало точности — уменьшаем шаг, увеличиваем разрядность числа… в общем боле менее методы понятны. Я же спрашиваю о том откуда при решении уравнения Шредингера возникает «экспоненциальная стена».
Victor_koly
15.12.2018 17:23По честному, в КМ вышло найти решение не очень большого числа практических задач (может что-то роустил):
1. Потенциальная яма (прямоугольная проще всего) и система ям, разделенная конечными барьерами.
2. Гармонический осциллятор.
3. Атом водорода. Магнитное поле ядра, спин-орбитальное взаимодействие, релятивистские эффекты, конечный размер ядра — это все приближения. Можно оценить уровень энергии, сложнее — найти новые волновые функции как лин. комбинацию функций базовой задачи.aslepov78
15.12.2018 17:34По честному и в любом другом разделе физики аналитических решений раз два и обчелся. Речь конечно о численном решении.
geisha
14.12.2018 23:53+1Симулировать — это, для начала, посчитать матричные элементы уравнения Шредингера. Количество матричных элементов — это размер вашего пространства векторов (в квадрате, что не так важно). Ключевой вопрос: какой размер у пространства векторов? Пусть вы хотите симулировать электрон в одномерной коробке (т.е. интервале). Вы, к примеру, разделяете интервал на 10 отрезков. Получается 10 векторов и 10х10 = 100 матричных элементов. Размер пространства — 10. Это легко симулировать.
Теперь вы добавляете второй электрон. Он тоже ограничен 10ю интервалами. В классической механике мы бы просто считали количество электронов в каждой точке и бед бы не знали. Но квантовая механика отвечает на вопросы типа «какова вероятность нахождения электрона 2 в одном из 10 подинтервалов при условии, что электрон 1 находится в интервале x?». Т.е. для каждого из x=1..10 случаев нахождения первого электрона есть 10 чисел (амлитуд), которые «вынь да полож». Этим амплитудам соответствуют новые вектора. Их 100 штук. Мы их называем состояниями: «состояние, при котором электрон 1 находится в подинтервале 1 и электрон 2 находится в подинтервале 1», и так далее в двойном цикле. Соответственно считать нам, в общем случае, порядка 10000 матричных элементов для двух электронов. Для трёх количество увеличится до 1e6 (а цикл будет тройным), для четырех — до 1e8 (четыре вложенных цикла) и т.д. Память у среднестатистического компьютера в этом случае закончится примерно на пятом-шестом электроне. (здесь тонна нюансов)
Это — т.н. экспоненциальная стена. Трагедия с таким ростом сложности состоит в том, что может оказаться, что ур. Шредингера для N электронов вы на лаптопе решаете, а для N+1 уже не существует суперкомпьютера, в который бы влезла волновая функция. Всё зависит от того, на сколько отрезков вы разбили интервал вначале. Если бы вы разбили его на минимально возможное количество отрезков — два, то ресурсы среднестатистического компьютера закончились бы где-то на 18м электроне, что тоже не ахти как много.
Может напишу статью по мотивам (а может и нет).geisha
15.12.2018 00:54+1На тот случай, если статью не напишу: все эти проблемы можно продемонстрировать и вне квантовой механики. Задача:
В пруду от берега к берегу растут N лилий в ряд. По лилиям прыгают жабы. Жабы прыгают между соседними лилиями и только в одну сторону. Для простоты, жабы прыгают в такт. Но вероятность того, что жаба перепрыгнет равна p в случае если лилия пуста и ноль, если на ней уже сидит жаба. С берега и на берег жабы прыгают с той же вероятностью при тех же условиях (считается, что на одном из берегов всегда есть жабы в достаточном количестве, а на другом — их нет). Какова средняя скорость движения жаб в зависимости от N, p?vassabi
15.12.2018 14:34Какова средняя скорость движения жаб в зависимости от N, p?
«методом Монте-Карло»: запускаем жаб для заданных N и p, считаем количество жаб, пересекших пруд и после 100500го раза смотрим на среднее, не?geisha
16.12.2018 02:39Можно. Но за 100500 раз вы никак не исследуете все конфигурационное пространство. В связи с этим возникает вопрос: а правильное ли среднее?
vassabi
16.12.2018 03:06Если дисперсия значений достаточно хорошо убывает при достижении 100500го шага на 100-м или 500-м запуске, то я буду склонен считать его скорее правильным, чем неправильным.
geisha
16.12.2018 05:42И, при всём этом, среднее окажется неправильным. К примеру, в случае, когда конфигурационное пространство разделено на две области равных размеров, причем из первой во вторую можно перескочить (но с достаточно малой вероятностью), а из второй в первую — нет. Вы просто будете получать одно из двух средних с правильной дисперсией.
vassabi
16.12.2018 14:34эхх! да, про случай с разными областями в которые разные средние, я и забыл.
Все время забываю, что на вопрос «какая АБВГ» у меня в голове скаляр, а у физиков в голове функция (или даже тензор) по (часто странному) пространству…
KonkovVladimir
16.12.2018 08:10Вы упустили один существенный шаг «метода Монте-Карло» — вам неизвестна равновесная конфигурация жаб и приходится начинать c неравновесного состояния. Для достижения равновесия вам нужно
исследовать все конфигурационное пространство.
как справедливо заметил(а) geisha — если цепочка лилий представляет произвольный замкнутый граф равновесную конфигурацию придется искать оч. долго.
Причем в квантовой химии часть жаб скачет спиной вверх, а часть спиной вниз при этом функция распределения должна быть строго антисимметрична относительно перестановки «двух разных спин», это существенно усложняет поиск равновесного состояния в этом веселом кардебалете.vassabi
16.12.2018 14:30Причем в квантовой химии часть жаб скачет спиной вверх, а часть спиной вниз при этом функция распределения должна быть строго антисимметрична относительно перестановки «двух разных спин»
АААААА! (убегает)
PS: после таких моментов так и вспоминается фраза Д.Мермина, я бы так наверное не смог бы…
aslepov78
15.12.2018 17:17Вы привели пример уже чисто математической задачи, в которой от физики вообще ничего не осталось. Я вижу что в квантах сплошь и рядом говорят о вероятностях, переходах, матрицах… Другое непонятно — как из уравнения Шредингера, которое непрерывное, в частных производных, причинно следственное (2го порядка)… и вдруг возникает вот эта задача на тему переборов и экспоненциального взрыва. И в тоже время в других областях, тоже с уравнениями в частных производных, тоже большая размерность — и вроде все в порядке, решаемо, нет таких принципиальных проблем.
geisha
16.12.2018 00:25Уравнение Шредингера записано для одной функции многих переменных: ?(x1, x2, x3, x4, ...) и в этом его главная сложность. Для классичеких полей вы записываете уравнение для, скажем, ?(x) и оно действительно проще потому что там меньше переменных. Про другие области не в курсе, приводите примеры.
Victor_koly
15.12.2018 00:04+1вплоть до области ?-квантов
Хотя формально гамма-кванты и рентген пересекаются по энергии, все же до диапазона гамма-квантов я бы не доводил даже уровни типа «энергия ионизации K-оболочки менделевия».
Тут нужно уточнить, что гамма-квантами с определенного уровня энергии называют отдельные кванты типа:
— энергия ионизации электрон-позитронной пары;
— энергия ядерного перехода нуклона в возбужденном ядре (тут она может быть пару МэВ).
В то время, как рентгеном называют частные случаи таких непрерывных процессов (легко объяснимых классической теорией поля):
— синхротронное излучение (но боюсь, что на LEP это были самые что ни есть гамма-лучи);
— тормозное (просто другой вид ускоренного движения.
KonkovVladimir
15.12.2018 17:35Безусловно полезно моделировать геометрические параметры отдельных молекул или периодических структуры, но с практической точки зрения так-же полезно моделировать другие св-ва соединений — плотность, температура плавления и кипения, твердость, UV-VIS спектр (цвет), например плотность воды рассчитанная разными методами (статья старая, но картинка нравится).

Взаимосвязь с реальностью тут гораздо более тесная, но и методы более изощренные и «трудоемкие».
Экспериментально установлено, что «зеленка» зеленая, алмаз самый твердый природный минерал, а YBCO переходит в сверхпроводящее состояние при 77K.
А что говорят об этом квантово-химические расчеты?
Вам не кажется, что квантово-химическая система (атом, молекула, периодическая структура) изоморфна квантовому компьютеру, по крайней мере существуют преобразования Jordan-Wigner или Bravyi and Kitaev переводящие одно в другое, а поскольку моделирование квантового компьютера на классическом задача заведомо тупиковая, ab initio квантово-химические расчеты на классических компьютерах ограничиваются простыми или модельными задачами в ожидании появления квантовых процессоров?madschumacher Автор
15.12.2018 18:49А что говорят об этом квантово-химические расчеты?
Квантово-химические расчёты говорят много чего. И UV/Vis спектры предсказываются, и ИК (включая и VCD), и даже масс-спектры!
То, что я писал только о структурах, так это из-за того, что я работаю в этой области, и из неё мне проще тырить примеры. Но в целом, данная статья распространяется и на другие экспериментальные методы исследования.
ab initio квантово-химические расчеты на классических компьютерах ограничиваются простыми или модельными задачами в ожидании появления квантовых процессоров?
Нет, не кажется. Благодаря многоуровневым методам (иерархической лестнице различных упрощающих предположений/грязных хаков) мы уже сейчас можем моделировать очень сложные системы. От сложных фотохимических процессов в биологических системах до разных каталитических процессов.
Вообще, с появлением новых компьютеров в квантовой химии сначала произошло некоторое атрофирование мозгов исследователей: вместо поиска каких-то элегантных путей решения задачи, они стали тупо запихивать всё внутрь компьютера, произносить некоторые «магические заклинания», которым они научились у Старинных Мудрецов, и ждатьчударезультата расчётов.
Сейчас же, когда пообвыклись с новыми возможностями, стали немного думать перед расчётом, из-за чего стало возможным «атаковать» более сложные и жизненные системы.
К чему это я? Чем ждать чуда у моря, лучше уже сейчас стараться и работать. :-)KonkovVladimir
15.12.2018 20:52Сейчас же, когда пообвыклись с новыми возможностями, стали немного думать перед расчётом, из-за чего стало возможным «атаковать» более сложные и жизненные системы.
Судя по статье в Nature Communicationsvolume в теории функционала плотности «думать человеческой головой» уже не получается — в ход пошла тяжела артиллерия в виде machine learning.
Один из авторов статьи, кстати, Kieron Burke — придумал в свое время (с друзьями), на мой взгляд, лучший DFT функционал всех времен и народов — на деле доказал, что «умел думать головой».madschumacher Автор
15.12.2018 21:03Один из авторов статьи, кстати, Kieron Burke — придумал в свое время (с друзьями), на мой взгляд, лучший DFT функционал всех времен и народов — на деле доказал, что «умел думать головой».
Вообще я слушал на одной из конференций доклад Бурка на тему этого направления (в частности он и об этой статье говорил). Там основная фишка подобных работ не сколько в повышении точности (PBE, где «B» is for «Burke», или B3LYP и так неплохи), а в ускорении. Просто всё равно все DFT методы масштабируются как O(M4)-O(M5) в зависимости от размера базиса (M), и поэтому посчитать ту же молдинамику для системы из сотен атомов в DFT не получится. А хочется…
Поэтому предполагается провести несколько дорогих расчётов, сделать аппроксимацию (то бишь обучить какую-ть нейросеть), а потом со спокойной душой и совестью пользоваться результатами этих аппроксимаций.
Это как раз один из возможных «грязных хаков», о которых я писал выше, чтобы начать моделировать реальные большие системы.geisha
17.12.2018 01:06Просто всё равно все DFT методы масштабируются как O(M4)-O(M5) в зависимости от размера базиса (M),
DFT — в подавляющем большинстве O(M3), инфа 100%. O(M5) — я вообще не в курсе о таких дорогих методах в DFT. Это сложность MP2 (т.е. не самого MP2, а поворота кулоновских интегралов).madschumacher Автор
17.12.2018 01:47geisha, спасибо за исправление-уточнение. Перепроверил приведённые цифры по F. Jensen «Introduction to Computational Chemistry» (2007-го года).
DFT — в подавляющем большинстве O(M3), инфа 100%.
При использовании разных «density fitting» техник (типа resolution of identity), да. Но стандартно (т.е. без удешевляющих технических приблуд), из-за наличия кулоновских интегралов

стоимость DFT сопоставима с Хартри-Фоком (т.е. O(M4)).
O(M5) — я вообще не в курсе о таких дорогих методах в DFT. Это сложность MP2 (т.е. не самого MP2, а поворота кулоновских интегралов).
Double-hybrids (т.е., по-сути, просто прикрученный MP2 к DFT), RPA (насчёт него не совсем уверен), и всё остальное, что влезет в последнюю ступень Лестницы Якова от Perdew?geisha
17.12.2018 02:45Кулоновское отталкивание <ij|ij> считается за O(M2). Это обменное взаимодействие <ij|ji> считается за O(M4). Конкретно PBE — O(M3) из-за диагонализации матрицы.
madschumacher Автор
17.12.2018 03:57geisha
F. Jensen «Introduction to Computational Chemistry» (2007-го года), раздел 6.8, стр. 261-262:
With an expansion of the orbitals in basis functions, the number of integrals necessary for solving the KS equations rises as Mbasis4, owing to the Coulomb integrals in the J functional (and possibly also “exact” exchange in the hybrid methods). The number of grid points for the numerical Exc integration (eq. (6.61)) increases linearly with the system size, and the computational effort for the exchange–correlation term rises as GM2 basis, i.e. a cubic dependence of the system size. When the Coulomb (and possibly “exact” exchange) term is evaluated directly from integrals over basis functions, DFT methods scale formally as Mbasis4.
Nevertheless, the formal Mbasis4 scaling has spawned approaches that reduce the dependence to Mbasis3. This may be achieved by fitting the electron density to a linear combination of functions, and using the fitted density in evaluating the J integrals in the Coulomb term.
Может я что-то не понимаю в этом куске?
Кулоновское отталкивание <ij|ij> считается за O(M2). Это обменное взаимодействие <ij|ji> считается за O(M4).
Тензоры кулоновского и обменного интегралов в представлении одинаковых базисов (пусть, условном базисе АО) имеют одинаковый размер. Почему они должны вычисляться за разное время? Если использовать разные схемы ускоренного расчёта (типа RI) для одного из этих кусков — понятно. Но это уже подразумевает дополнительные условия. Нет?
P.S. А если не секрет, откуда эта инфа 100%?
saga111a
15.12.2018 21:51А подробнее можете сказать какие данные в CSD и что нужно для получения базы/доступа?
Требуется ли «институтская почта»?madschumacher Автор
15.12.2018 21:58А подробнее можете сказать какие данные в CSD и что нужно для получения базы/доступа?
Это сборник кристаллических структур + встроенный вьюер структур + простейший поиск по ним. Структуры только для различных молекулярных кристаллов. Можно даже скачать эти данные, они хранятся в формате *.cif. Только для скачивания нужно ввести свою почту.
Требуется ли «институтская почта»?
Нет, емнип, институтская не нужна, подойдёт любая. Но вот место работы нужно будет заполнить (в опроснике). Система похожа на то, что теперь требует УФН. Не строго, никакого контроля, но извольте заполнить.
Вообще, с CSD, чем 100 раз прочитать, лучше 1 раз попробовать. ;)saga111a
15.12.2018 22:23А вот из тех «Решений» кто конкретно подразумевается под бесплатными?
CSD-System?madschumacher Автор
15.12.2018 22:41Поскольку она предполагает допуск к ICSD, не уверен.
Вот ссылка на заведомо бесплатный поиск:saga111a
15.12.2018 22:50веб поиск я находил, а вот там много где писали что их базу можно скачать, а конкретно что скачивать не указано)
saga111a
15.12.2018 23:29Я вот в CSD-system не вижу про ICSD ничего.
к словуК ICSD доступ имею, но только на рабочей «машине».
COD база хорошая но органики там нету, как назло =( может у CSD она нормальная.
PDF-2 2004 имею в доступности, но она старая как черт и там много чего нету.madschumacher Автор
16.12.2018 15:08На странице
More advanced search functionality and additional curated data for the Cambridge Structural Database (CSD) and the Inorganic Crystal Structure Database (ICSD) is available through the CSD-System and ICSD, respectively. Click here for more information.
Я вчера просто невнимательно прочитал. По ссылке в описании юзер просто перенаправляется на саму ICSD.
может у CSD она нормальная.
В CSD органика точно есть, но выгрузить её можно только по одной (если нет платного доступа).
DustCn
17.12.2018 04:55Интересно было бы в сторону различного софта углубить и расширить статью.
В силу свой рабочей специальности как программист ковырялся в потрохах разных MD кодов, хотелось бы заслушать людей, скажем так, с той стороны баррикад.
Кроме приснопамятного Гауссиана есть же еще много интересных пакетов. CP2K упоминался уже, который выделяется тем, что хранит в хитром блочном «а-ля» спарс формате, правда по этому проседает по скорости. Ну или QuantumEspresso бесплатный, основанный на CASTEP, который к тому же очередь прародитель VASP-а.
madschumacher, может черканете пару абзацев на такую тему? Скорость, точность, устойчивость рассчетов, и тп… Интересно же, для нас это просто строчки кода :)madschumacher Автор
17.12.2018 10:32Скорость, точность, устойчивость рассчетов, и тп… Интересно же, для нас это просто строчки кода :)
К сожалению, тут я не очень большой специалист, т.к. я сам работал только с Gaussian, GAMESS US/Firefly, Orca (в порядке истории) + совсем немного Priroda и CP2K (но на уровне шапочного знакомства с расчётами). Поэтому тут я ничего особо полезного сказать не могу, к сожалению.
Единственное, что могу сказать, это что Турбомолевский юзеровский интерфейс нарушает все возможные права человека, и что он должен быть запрещён отдельной конвенцией ООН (правда по производительности и возможностям — офигенный код, стоящий своих денег).DustCn
17.12.2018 12:37К сожалению когда ученый-прикладник осеняется знамением и решает написать свой супер-турбо-DFT на стероидах обычно получается что то невнятное по интерфейсу. И если наша братия подхватив это знамя путем рефакторинга бывает спасает ситуацию, то для закрытых кодов шансов почти нет :)
Вам повезло что вы еще внутрь не заглядывали. Почти все MD кода это Фортран, из С/C++ припомню разве что LAMMPS.
R9A_019
Шикарная статья, но у меня вопрос, вот там у вас "схема вводимых приближений" и от релятивизма и прочих радостей, сразу отказываются. А поправки от них вниз по схеме распространяются? или уравнение Дирака с прочими радостями, не существенно влияют на получаемые дальше модели (понятно что все зависит от той системы для которой модель и строится, но все таки)?
madschumacher Автор
Релятивизм является существенным только для электронов в тяжёлых элементах (4й период Периодической таблицы и далее). В лучшем случае достаточным окажется только модифицировать поверхность потенциальной энергии (т.е. учесть релятивизм электронов), тогда остальная схема не сломается.
Но, вполне вероятным может оказаться вариант, когда учёт релятивизма у электронов может сломать приближение Борна-Оппенгеймера. Тогда вся остальная схема накроется медным тазом, и придётся переходить к более сложным моделям.
P.S. уравнение Дирака при этом обычно не используют, юзают релятивистские поправки к Шрёдингеру.