

В настоящее время сложно кого-то удивить терминами «ген», «геном», «ДНК», настолько плотно они вошли в нашу повседневную жизнь. Все слышали о том, что геном человека был расшифрован, однако немногие из нас четко понимают значение этого научного прорыва для всего человечества. Исправление человеческих «слабостей», увеличение продолжительности жизни и нахождение все более эффективных способов борьбы с преступностью – все это становится возможно благодаря изучению наследственной информации, содержащейся в геноме человека.
Немного теории
Поскольку большинство из нас несколько подзабыло школьный курс биологии, предлагаем освежить в памяти основные понятия:
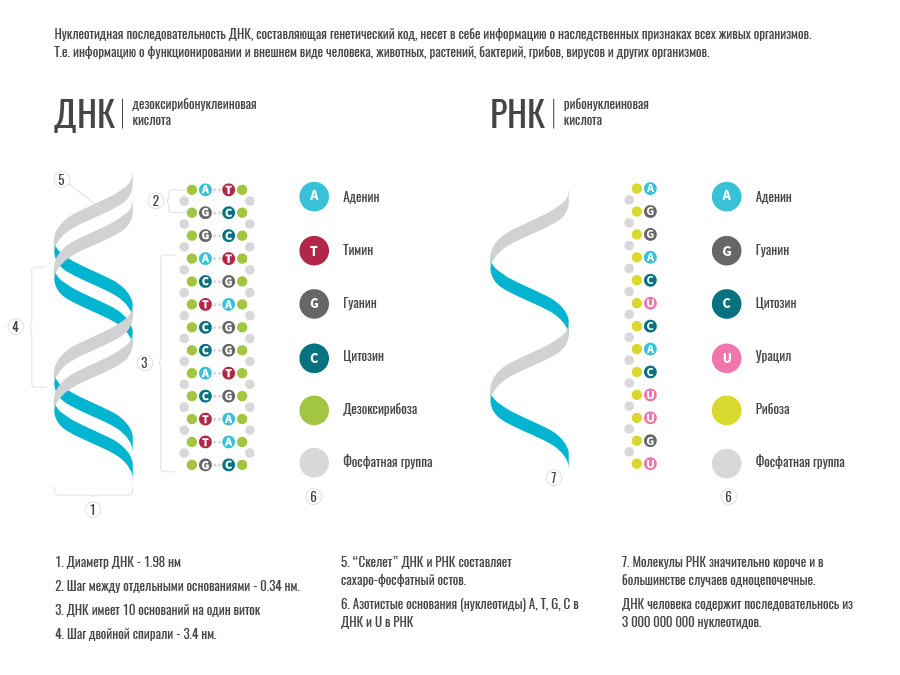
Дезоксирибонуклеиновая кислота, более известная, как ДНК, — высокополимерное природное соединение, содержащееся в ядрах клеток всех живых организмов, носитель генетической информации. ДНК состоит из 4 видов нуклеотидов. Основу нуклеотидов ДНК составляет сахар дезоксирибоза (одинаковый для всех нуклеотидов) и 4 типа азотистых оснований — аденин (A), тимин (T), гуанин (G) и цитозин ©. Именно последовательность нуклеотидов и определяет записанную в ДНК информацию. Отдельные участки ДНК соответствуют определенным генам.
Ген, в свою очередь, — это физическая (определенный участок ДНК) и функциональная (кодирует белок или рибонуклеиновые кислоты) единица наследственности. Важнейшее свойство генов — сочетание их высокой устойчивости в ряду поколений со способностью к наследуемым изменениям (мутациям), служащим основой изменчивости организмов, дающей материал для естественного отбора.
Рибонуклеиновые кислоты (РНК) — тип нуклеиновых кислот, которые состоят из 4 нуклеотидов, построенных на основе сахара рибозы и азотистых оснований – это аденин, гуанин, цитозин, урацил (A, C, G, U). Урацил (U) является полным аналогом тимина (T) в ДНК, так что для наших целей можно считать их идентичными. В клетках всех живых организмов РНК участвуют в реализации генетической информации. С находящихся в ДНК генов создается РНК-копия, которая либо функционирует сама по себе, либо служит матрицей для синтеза белка.
Что же такое геном? Геном – это ДНК, содержащаяся в гаплоидном наборе хромосом клетки определенного вида организма. В расширенном виде под геномом понимается вся наследственная система клетки.
Про 23 пары хромосом в геноме человека, думается, помнят все. Первоначально хромосомами назвали структуры из очень плотно свернутой нити, которая образуется в клетке в определенный момент в процессе деления, и видна в обычный оптический микроскоп. Хромосомой называют и саму нить ДНК в «расправленном» состоянии.
Рид — отдельное прочтение фрагмента ДНК. В свою очередь, локус – это местоположение определенного гена на генетической или цитологической карте хромосомы.
Геном и медицина
Про «расшифровку» генома человека так или иначе слышали все. К сожалению, сам термин «расшифровка» выбран неудачно, что постоянно вызывает вопросы. Правильно сказать, что геном человека был прочитан (секвенирован), т. е. получена полная последовательность нуклеотидов всех нитей ДНК.
Чтение ДНК — это сама по себе очень сложная техническая задача. Но даже после прочтения такой геном предстает нам в виде строки, состоящей из четырех букв (A, C, G, T), причем его длина может достигать нескольких миллиардов символов. При этом в ДНК нет абсолютно четких «знаков препинания», которые бы указывали на начало и конец генов-предложений и других функционально значимых элементов. Эта огромная по объему информация чем-то похожа на полный дамп памяти компьютера. А понять по этому дампу, как наш живой компьютер работает — отдельная, значительно более сложная задача.
Раньше для анализа были доступны только короткие кусочки последовательности ДНК — отдельные гены и их фрагменты. Благодаря полному секвенированию генома стало намного легче анализировать «текст» ДНК и искать в нем функциональные участки. За последние 10 лет ученые очень сильно продвинулись в этом направлении. Были обнаружены новые гены, мутантные формы которых приводят к раку, атеросклерозу, болезни Альцгеймера, сердечно-сосудистым заболеваниям. Предрасположенность человека к алкоголизму, наркомании, азартным играм, психическим заболеваниям и даже суициду имеет генетическую основу. Более того, склонность к поиску новых впечатлений, материнский инстинкт, агрессивность поведения, активность и раздражительность также находятся под жестким генетическим контролем! Конечно, не всякая заложенная в ДНК возможность будет реализована, но именно гены задают нам набор возможностей и рисков, на основе которых строится наша жизнь.
К слову, травмирующий опыт родителей может напрямую передаваться потомкам через так называемое эпигенетическое наследование. Страхи и стрессы предков значительно влияют на структуру и функцию нервной системы последующих поколений. И если вы непонятной причине боитесь собак, возможно, кого-то из ваших предков когда-то покусали. Эта область еще изучена очень мало, но также активно развивается.
ДНК-тест позволяет обнаружить предрасположенность человека к тем или иным заболеваниям, в том числе, и обусловленным внешними причинами (вирусы, питание и т.д.), идентифицировать личность, установить отцовство, определить совместимость донора и реципиента при проведении трансплантации и многое другое.
Бывает и такое. В 2002 году американка Лидия Фэйрчайлд при разводе с мужем проходила процедуру анализа ДНК, которая показала, что она не является матерью двум её детям. В то время она уже была беременна третьим, пробы крови которого после рождения показали, что она не является матерью и ему. Дальнейшие исследования выявили, что Лидия — химера, то есть организм, обладающий двумя различными геномами.
Геном и биоинформатика
Полное секвенирование генома отдельного человека позволяет одномоментно получить полный текст ДНК и провести анализ сразу всех его генов вместо нескольких тысяч специализированных тестов на отдельные наследственные заболевания. Проект «Геном человека» потребовал 5 лет и несколько миллиардов долларов. Современные машины позволяют полностью секвенировать геном человека за 15-20 дней по цене около $1000/геном. Тогда почему же исследование геномов и геномная диагностика происходят не так быстро, как хотелось бы?
Дело в том, что ни один современный секвенатор не может прочесть всю нить ДНК от начала до конца. Для секвенирования ДНК случайным образом нарезают на короткие фрагменты, которые секвенатор затем параллельно читает в больших количествах. В результате получаются файлы размером в сотни гигабайт, в которых находится колоссальное количество коротких (150-300 нуклеотидов) прочтений.
Поскольку в одном биологическом образце много клеток, а ДНК фрагментируется случайно, прочитанные фрагменты будут многократно перекрываться.
Для получения нужного генома необходимо собрать этот паззл в полный текст. Это колоссальная вычислительная задача, и обработка таких файлов занимает продолжительное время. Все данные обрабатываются специальными геномными ассемблерами, которые и собирают максимально продолжительные по длине участки генома.
EPAM, собери мне геном
Расхожая истина, что время – деньги, верна и для геномных исследований. Расшифровка наследственного материала открывает фантастические возможности для медицины: от «ремонта» дефектных генов до нахождения тайны вечной молодости. И именно поэтому множество научных институтов и частных компаний прилагают невероятные усилия для ускорения процесса обработки генома.
Для обработки и анализа результатов сиквенса создано большое число программ. Однако производительность секвенаторов и объемы получаемой информации растут с такой скоростью, что разработчики часто не успевают оптимизировать свои программы для обработки все увеличивающегося количества данных. Возникает потребность адаптировать разработанные программы для параллельной работы, использования вычислительных кластеров и т.д.
Один из клиентов обратился к нам за автоматизацией процесса контроля качества сборки генома и сокращением вычислительного времени.
В данный момент мы оптимизируем различные процессы, чтобы минимизировать время и, соответственно, затраты. Работа идёт с небольшими утилитами, которые занимаются расчетом метрик — численных характеристик, по которым экспериментатор контролирует качество подготовки образца, проведения секвенирования и сборки генома. Метрики позволяют оценить тот файл, который выдается секвенатором: подходит он для дальнейшего анализа генома или же нужно провести еще один эксперимент.
Метрики и алгоритмы – оптимизация работы
Благодаря работе нашего отдела алгоритмов, время расчета одной метрики сократилось с 11 часов до 105 минут. Раньше четыре метрики серьезная машина обсчитывала за 25 часов. Мы, с помощью многопоточности и переработки алгоритмов, сократили это время в четыре раза. И это не предел.
Оптимизация метрик заключалась в поиске точек в коде для ветвления процесса вычислений. Все метрики разные по структуре, но одна общая черта у них есть: все они читают файл в формате .bam, в котором хранятся данные об отдельных прочтениях фрагментов ДНК (ридах).
Схема оптимизации проста: обработчики данных используют итератор, который читает из файла по одной записи (SAMRecord), и производят анализ информации с накоплением промежуточных результатов (статистики) в неких структурах данных.
Одна из оптимизированных метрик позволяет собирать статистику из .bam-файла, с помощью которой можно оценить, насколько качественно был проведен эксперимент. Чтобы отличить ошибку секвенирования от мутации, в статистике должно быть достаточно большое количество отдельных прочтений фрагментов ДНК (ридов). Очень большое покрытие говорит о том, что эксперимент можно было провести дешевле без потери качества. В итоге метрика выдает информацию о том, сколько выровненных оснований было отфильтровано из-за низкого качества.
В чем же состоит оптимизация? Первоначально алгоритм считал статистику для каждого месторасположения определенного гена (локуса), проходя по всем ридам, покрывающим эту позицию. Если рид состоял из 300 оснований (а это его стандартная длина), то алгоритм 300 раз считывал этот фрагмент ДНК, чтобы получить информацию о качестве его основания. Алгоритм, оптимизированный одним из наших специалистов, считывает данные о качестве всех оснований в тот момент, когда рид встречается первый раз, что позволяет избежать обращения к нему для каждого локуса. Информация об отфильтрованных основаниях накапливается в массиве-счетчике, который сдвигается по мере прохождения по геному. Таким образом, статистика по каждому прочтению собирается только один раз, что позволяет существенно ускорить процесс обработки данных.
Ещё одна метрика собирает статистику о дубликатах (т.е. идентичных ридах, совпадающих до заданного числа оснований). На основе этой статистики оценивается сложность библиотеки ридов. Слишком высокое количество дубликатов показывает, что эксперимент можно было провести дешевле. Низкая же сложность библиотеки говорит о том, что мы имеем дело с маленьким количеством прочтений, что может исказить точность результатов при дальнейшей работе с геномом.
Изначальный алгоритм разбивал риды на подмножества, объединяя их по первым n-элементам. Далее происходило сравнение прочтений фрагментов ДНК для каждого из них. Очевидно, что такой алгоритм работает крайне медленно (для больших подмножеств время растет экспоненциально). Оптимизацией в данном случае является использование модифицированного LSH-алгоритма (locality-sensitive hashing algorithm). Каждый рид разбивается на шинглы, то есть небольшие кусочки, включающие в себя несколько оснований и позицию начала этого кусочка в риде. Далее строится таблица, в которой хранится информация о том, в каких ридах встречается этот шингл. Затем с помощью случайных перестановок определяются похожие прочтения, которые сравниваются посимвольно. Использование данной оптимизации позволяет избежать огромного количества ненужных сравнений.
Многопоточность
Как уже говорилось выше, наши специалисты смогли значительно сократить время просчета метрик. Это стало возможно не только за счет переработки алгоритмов, но и с помощью многопоточности. Какие же изменения были сделаны?
Был создан отдельный поток для чтения записей из .bam-файла, который накапливает их в отдельном кольцевом буфере (bounded buffer). Как только буфер заполняется, он складывается в ограниченную очередь (bounded queue). И буфер, и очередь таких заполненных буферов ограничены из соображений конечного объёма выделяемой процессу оперативной памяти.
Другой поток (или несколько потоков для некоторых метрик) берет очередной полный буфер из этой очереди и использует его в качестве источника данных для итератора. Далее все действует так же, как и в первоначальной схеме.
Накопление результатов обработки записей из нескольких потоков требует применения новых структур данных для статистики метрик. Делается это для того, чтобы избежать состояния гонки (race condition). С этой целью мы создали дополнительные классы для накопления статистики и применяем атомарные операции (atomics).
Атомарные операции приводят к относительно высоким затратам на консолидацию промежуточных статистических данных из разных потоков для отдельных метрик. Для того, чтобы ускорить процесс обработки, записи были сделаны в виде блоков по несколько сотен в одном потоке.
Благодаря проведенным оптимизациям, время просчета метрик сократилось в несколько раз. Тем не менее, это не предел. Для отсортированных индексированных .bam-файлов возможен переход к параллельному чтению и обработке данных по отдельным хромосомам, что позволит ещё больше сократить время обработки данных генома.
Напоследок
О генетике и геноме можно говорить практически бесконечно, благо тема интересная и касается всех нас. Возможно, благодаря постоянной работе над автоматизацией обработки генома уже через пару лет его расшифровка и сборка будут занимать буквально несколько часов, а стоимость снизится настолько, что абсолютно каждый человек сможет позволить себе провести генный анализ. Фантастика? Вряд ли. Геномные технологии сейчас развиваются почти по закону Мура, производительность секвенаторов удваивается примерно каждые 2 года. Поэтому вполне вероятно, что за следующие 10-15 лет геномные технологии станут настолько же распространенными и привычными, как уже стали смартфоны и ноутбуки.
P.S. В силу ограниченного объема статья не претендует на строгую научную правильность в описании биологических терминов и процессов.
Комментарии (18)

Megakazbek
04.05.2015 16:05+2Интересно, что это за молекула изображена на заглавной картинке и какое она отношение имеет к ДНК.
ninch
04.05.2015 23:47Пожалейте копирайтеров, в интернете всего три с половиной модно выглядящих рендёра двойной спирали.

Londoner
04.05.2015 20:31Современные машины позволяют полностью секвенировать геном человека за 15-20 дней по цене около $1000/геном.
А как же тогда 23andme умудряется это делать всего за $99?

ninch
05.05.2015 00:01+1Дубликаты — вовсе не идентичные риды. Это независимо прочтённые риды с одного и того же ампликона. Они могут отличаться друг от друга по качеству и даже по контенту (ошибки ПЦР никто не отменял).
Слишком большое количество дублей говорит вовсе не о том, что эксперимент можно было бы провести дешевле, а наоборот — что на нём продешевили (даже если потратили больше денег, чем достаточно для того же уровня итогового качества). Плохой реагент-кит, неправильно порезали, слишком много циклов ПЦР, наконец, сингл-риды вместо парных библиотек.

m08pvv
05.05.2015 00:26А когда наступит момент, когда можно будет собрать, по аналогии с искусственной рибосомой, что-то, что будет в живой клетке вычитывать ДНК и генерировать что-то, что проще прочесть и долговечнее? Или это принципиально невозможно/сложно/дорого?

dmitrmax
05.05.2015 21:46+1> При этом в ДНК нет знаков препинания, которые бы указывали на начало и конец генов и других функционально значимых элементов.
Я не специалист в этой области, но, по-моему, вы здесь слукавили. Из доклада по биологии в школе помню, что есть старт- и стоп-кодоны (AUG и UGA) соответственно, кроме того, есть некоторые последовательности, которые маркируют начало и конец интрона, внутри гена.
Aksinnia Автор
07.05.2015 14:31Спасибо за замечание, поправила в тексте, чтобы было более понятно, что имеется в виду. В ДНК нет абсолютно чётких «знаков препинания», которые бы ясно давали понять, что вот здесь вот начало, а здесь конец гена. В ДНК нет химических модификаций или особенных букв (не ATGC), которые маркировали бы функциональные единицы. Из-за этого и возникают проблемы. Далеко не каждый AUG является стартом трансляции белка, они часто встречаются и в межгенных промежутках. Начало и конец интрона тоже маркируются, но такие же последовательности опять встречаются и вне белок-кодирующих генов, так что в случае, если не известны начало и конец гена, эта информация не имеет значения. А маркировки других функциональных элементов – сайтов посадки рибосомы, промотора гена, сайтов посадки сопутствующих белков часто намного более сложные и менее очевидные.
Протяженные сигналы не обязаны повторяться буква в букву. Например, промотор гена у бактерий может содержать замены, которые ухудшают распознавание этого промотора полимеразами. Это один из способов снизить активность этого гена. В результате найти такой «испорченный» промотор становится сложнее.
Задача отыскания гена как раз и состоит в отыскании некоторой комбинации его возможных функциональных элементов. Т.е. если мы видим участок, в котором компактно собраны и стартовый кодон, и сайт посадки рибосомы, и промотор для РНК-полимеразы – скорее всего это ген. Но каждый из этих сигналов может быть неточным и содержать ошибки, так что задача весьма непростая.
И даже найдя ген надо еще понять, что он делает и зачем нужен.
guai
А насчет покусанных собаками пруфы будут?
FreeLSD
Думаю все проще.
Есть несколько особей, живущих в населенном агрессивными собаками районе.
У одной из особей появляется мутация, которая в том числе влияет на страх перед дикими животными. Эта мутация дает особи существенное конкурентное преимущество перед сородичами, активнее происходит размножение и ген быстро распространяется в популяции.
Aksinnia Автор
Эксперименты, проведенные учеными из университета Эмори над мышами, показали, что травмирующие события в жизни изменяют активность генов путем химической модификации ДНК, что вносит изменения в работу мозга и поведение последующих поколений. Оригинал исследования из журнала NATURE NEUROSCIENCE можно почитать здесь, если есть такая возможность. Обзорную статью (по-русски) на исследование можно посмотреть вот здесь.
clansman
Это говорит только о том что у мышей есть адаптация которая позволяет быстро передать страх перед запахом потомкам. Эта адаптация могла возникнуть в случае контакта с большим разнообразием токсинов и она полезна для выживания (так как однажды обнаруженная отрава не станет безвредной в скором времени) значит она преуспела в популяции. Но если говорить о человеческих страхах — во первых они сложней, думаю не так то просто по быстрому накодить страх перед чем-то похожим на собаку. Во вторых эта особенность не выглядит полезной. То что больших хищников надо бояться мы все знаем, но стоит мне пару раз облажаться с животным безвредным в большинстве случаев — мои дети получат лишнюю причину для беспокойства.
guai
Пока не читал инфу по ссылкам. Они там прям нашли изменившиеся, после травмирущих событий, гены?
Что-то мне кажется, в любом случае перенос этих данных на людей и собак, как в статье, окажется сильно натянутым…
hungry_ewok
/усмехаясь/
Тень Трофима Денисовича укоризненно качает головой, как бы намекая: «А я вам говорил!»