
Библиотека ASE
Библиотека ASE — это python-библиотека для проведения атомных манипуляций и вычислений. В данной статье мы будем создавать наночастицы с помощью этой библиотеки.
Установка
Устанавливается ASE стандартно через pip: pip install ase.
Объект Atoms
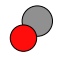
Основной объект, с которым можно проводить манипуляции — это объект Atoms. Он может хранить в себе различные структуры. Давайте попробуем создать молекулу угарного газа:
from ase import Atoms
from ase.visualize import view
d = 1.1
co = Atoms('CO', positions=[(0, 0, 0), (0, 0, d)])
view(co)В результате получим структуру следующего вида:
Кластеры
Создание кластера — частицы, имеющей кристаллическую решетку, — возможно с помощью модуля ase.cluster. Генерируются кластеры посредством отсечения от бесконечного кристалла кусков плоскостями отсечения. Создадим кластер меди:
from ase.cluster.cubic import FaceCenteredCubic
surfaces = [(1, 0, 0), (1, 1, 0), (1, 1, 1)]
layers = [6, 9, 5]
lc = 3.61000
atoms = FaceCenteredCubic('Cu', surfaces, layers, latticeconstant=lc)
view(atoms)где surfaces — плоскости отсечения, layers — число слоев.
Получаем структуру:

Наночастицы
И вот мы подходим к основной теме статьи. Наночастицы — это частицы порядка нанометровых размеров в диаметре (до сотен нанометров). Наиболее распространены сферические наночастицы — их мы и будем генерировать. Как это сделать? Давайте будем создавать большой кластер, а затем вырезать из него сферу — по удаленности координаты атома от центра. Для этого сперва необходимо центрировать все координаты:
surfaces = [(1, 0, 0), (1, 1, 0), (1, 1, 1)]
layers = [50, 50, 50]
lc = 3.61000
atomsCu = FaceCenteredCubic('Cu', surfaces, layers, latticeconstant=lc)
possCu = atomsCu.get_positions()
cCu = atomsCu.get_center_of_mass()
new_possCu = []
for el in possCu:
new_possCu.append(el - cCu)
new_possCu = np.array(new_possCu)Удаленность центрированного атома от центра рассчитывается просто:
def get_radius_centered(pos):
return (pos[0]**2 + pos[1]**2 + pos[2]**2)**0.5Теперь пишем функцию для вырезания из большого кластера нашей сферической частицы (точнее, их позиции):
def cut_atoms(poss, r):
poss_cut = []
for pos in poss:
if get_radius_centered(pos) < r:
poss_cut.append(pos)
poss_cut = np.array(poss_cut)
return poss_cutОсталось применить это на практике:
r = 5
possCu = utils.cut_atoms(new_possCu, r)
atoms = Atoms('Cu'+str(len(possCu)), positions=possCu)
view(atoms)Получаем следующее изображение наночастицы меди с радиусом 5 ангстрем (0.5 нм):

Core-shell структуры
На практике зачастую интересны core-shell структуры — с ядром одного типа атомов и оболочкой — другого. Например, в реальной жизни core-shell частицы CuAg (с ядром Cu) могут обладать сравнимой с Ag каталитической активностью, но быть при этом дешевле. Научимся генерировать такие структуры.
Будем придерживаться следующей стратегии: вырежем из частицы дырку и заполним эту дырку частицей с другого типа атомами. Для этого нужна функция, вырезающая дырку:
def cut_atoms_inside(poss, r):
poss_cut = []
for pos in poss:
if get_radius_centered(pos) > r:
poss_cut.append(pos)
poss_cut = np.array(poss_cut)
return poss_cutСоздадим кластер серебра с центрированными координатами атомов:
lc_ag = 4.09
atomsAg = FaceCenteredCubic('Ag', surfaces, layers, latticeconstant=lc_ag)
possAg = atomsAg.get_positions()
cAg = atomsAg.get_center_of_mass()
new_possAg = []
for el in possAg:
new_possAg.append(el - cAg)
new_possAg = np.array(new_possAg)и создадим core-shell частицу:
r1 = 7
r2 = 5
possAg = utils.cut_atoms(new_possAg, r1)
atoms = Atoms('Ag'+str(len(possAg)), positions=possAg)
possAg = utils.cut_atoms_inside(possAg, r2)
possCu = utils.cut_atoms(new_possCu, r2)
poss = np.vstack([possCu, possAg])
atoms = Atoms('Cu'+str(len(possCu))+'Ag'+str(len(possAg)), positions=poss)
view(atoms)Визуализация показывает, что мы добились того, чего хотели:

Теперь можем записать координаты в файл:
atoms.write('Cu'+str(r1)+'Ag'+str(r1-r2)+'.xyz')Релаксация
И, наконец, пример манипуляций, ради которых создавалась библиотека. Отрелаксируем core-shell частицу, т.е. приведем к состоянию с минимальной потенциальной энергией. Задаем калькулятор:
from ase.calculators.emt import EMT
atoms = Atoms(atoms, calculator=EMT())и запускаем релаксацию:
dyn = BFGS(atoms, trajectory='atoms.traj')
dyn.run(fmax=0.05)Конечное состояние, как видим, отличается от идеальной структуры выше:

Код доступен на гитхабе.