
В обновленном на прошлой неделе списке Всемирной организации здравоохранения числится уже 131 проект разработки вакцин, призванных сформировать иммунный ответ к вирусу SARS-CoV-2 — причине заболевания COVID-19. Среди них есть два проекта от нашего партнера — российской биофармацевтической компании BIOCAD. Попадание в этот список — не только вопрос престижа, но и возможность включиться в рабочие группы разработчиков вакцин по всему миру, обсуждающих без лишних глаз вопросы клеточных и животных моделей, на которых можно было бы протестировать те или иные вакцины. Тем не менее за пределы таких специализированных групп информация о том, как именно разрабатываются вакцины, проникает крайне скупо.
В СМИ активно раскручиваются обсуждения клинических исследований, производства и будущего применения вакцин, тогда как «ранние» разработчики, скорее, отмахивались от излишней публичности. Теперь же вся часть по дизайну и сборке кандидатов завершена, мы затаились в ожидании результатов с животных моделей и получения разрешений на людях. Так что можно выделить немного времени для того, чтоб рассказать какие задачи возникают внутри.
Поскольку Habr — ресурс в первую очередь околоайтишный, а в современной фармацевтике активно применяются методы биоинформатики и вычислительной биологии, мы остановимся именно на «сухих» математических задачах. На самом деле эти задачи выбраны еще и потому, что в следующем году НИУ ВШЭ — Санкт-Петербург и BIOCAD открывают свою магистратуру по вычислительной биологии, но об этом в самом конце. В первую же очередь разберемся, что, собственно, нужно было сделать.
Над этой статьей работали три сотрудника BIOCAD:
Диана Кондинская — выпускница биофизики физического факультета СПбГУ, старший специалист компании по структурной биоинформатике. Своими руками делает белковые дизайны оригинальных препаратов, занимается молекулярными симуляциями и всем, что с ними связано.
Александра Черткова — выпускница биоинформатики СПбГПУ. Занимает должность руководителя отдела системной биологии, отвечает за моделирование фармакокинетики и фармакодинамики всех доклинических и клинических исследований компании.
Павел Яковлев окончил программу по биоинформатике в Академическом Университете. В ходе выполнения первого же научно-исследовательского проекта в университете попал в BIOCAD, оказавшись там первым биоинформатиком. Последовательно прошел должности руководителя группы биоинформатики и директора департамента вычислительной биологии. С февраля 2020 года стал руководить всеми исследованиями в области разработки оригинальных препаратов на должности директора по ранней разработке и развитию.
Говоря простым языком, вакцины — это тип лекарственных препаратов, задача которых — «научить» иммунную систему находить и уничтожать определенные патогены. Традиционно при этом подразумеваются профилактические вакцины, которые тренируют иммунитет еще до попадания патогена в организм, но в некоторых областях бывают и исключения, например, терапевтические онковакцины.
Рассказывать в двух словах про работу иммунной системы высших млекопитающих — дело неблагодарное. Курс иммунологии на любом биофаке традиционно считается одним из самых сложных, да еще и устаревает обычно быстрее, чем идет семестр. Так что, ни в коем случае не претендуя на полноту, обратимся только к главным интересующим нас деталям адаптивного иммунитета, вырабатывающегося организмом индивидуально к новым угрозам.
Первым важным элементом является В-клеточный иммунитет. Его суть заключается в постоянном (на протяжении всей жизни!) образовании новых клеток, называемых В-лимфоцитами. Каждый такой лимфоцит способен распознавать уникальную мишень с помощью своего В-клеточного рецептора. Также эти клетки способны производить специальную форму этих рецепторов — иммуноглобулины (или антитела), которые не остаются на поверхности клетки, а плавают свободно, специфически связывая патогенные белки как в свободной форме, так и на поверхности клеток. Такие антитела способны не только заблокировать какое-либо взаимодействие, но и пометить клетку на уничтожение другими компонентами иммунной системы — фагоцитами и натуральными киллерами. Кстати, синтетические антитела являются основой для современной противораковой терапии.
Наличие В-клеточного иммунного ответа подразумевает некоторую встроенную в организм систему «свой-чужой», которая бы позволила идентифицировать внешние патогены и критичные мутации собственных белков и вызвать на них иммунную реакцию. Действительно, абсолютно каждая клетка нашего организма содержит систему, обеспечивающую презентацию производимых клеткой белков на поверхности. Так, специальный белковый комплекс, называемый протеасомой, способен расщеплять белки внутри клетки на короткие линейные фрагменты — пептиды, состоящие не более чем из пары десятков аминокислот. Такие пептиды могут связываться с продуктами аллелей HLA, кодирующих специальные белки MHC (в данном случае I типа). Несмотря на то, что этих аллелей в человеческом геноме заложено несколько тысяч (см., например, здесь), активно работают всего несколько сотен, при этом в каждом человеке свои. Это определяется как наследственностью, так и внешними факторами, так что обычно справедливо предполагать, что у жителей одного региона большая часть активных MHC будет одинаковой. Связываясь с продуктами протеасомной деградации, МНС I типа выходят на мембрану клетки, таким образом показывая продукты, вырабатываемые клеткой.
Помимо механизма презентации своих внутренностей каждой клеткой, существуют также профессиональные демонстраторы — антиген-презентирующие клетки. Они способны захватывать объекты из внешней среды и показывать их пептиды аналогичным образом, используя для этого уже MHC II класса, которые также являются продуктами HLA.
Но раз кто-то демонстрирует продукты, какая-то система должна их и распознавать. За это отвечает вторая важная ветвь адаптивной иммунной системы — Т-клеточный иммунитет. С помощью специальных рецепторов (называемых, очевидно, Т-клеточными) такие клетки способны распознавать пептиды, не соответствующие нормальным белкам организма и вызывать различные иммунные реакции. Клетки T-хелперы будут смотреть на комплексы из MHC II класса и презентируемых ими пептидов и активировать В-клеточный иммунитет и ряд других реакций. Цитотоксические клетки Т-киллеры просто будут уничтожать всех, чья пара из MHC I класса и презентуемого пептида им не понравится.
Вооружившись этим знанием, теперь мы можем осознать истинную задачу вакцины — обеспечить в организме наличие белков патогена (в нашем случае вируса SARS-CoV-2), чтобы запустить выработку В- и/или Т-клеточного иммунитета, который будет способен быстро нейтрализовать сам патоген при его попадании в организм. Вырабатываемые антитела будут препятствовать проникновению вируса в клетку, а Т-лимфоциты смогут убивать клетки, в которых вирус уже поселился.
Очень часто в качестве вакцин используют ослабленные патогенные микроорганизмы — вирусы и бактерии, которые несут на себе характерные распознаваемые иммунной системой белки, но сами по себе безвредны. Такие вакцины называются аттенуированными. Тем не менее, разработка и доказательство их безопасности — сложный и трудоемкий процесс. В качестве эффективной безопасной альтернативы можно использовать рекомбинантные вакцины, созданные с помощью генно-инженерных методов. В рекомбинантных вакцинах используются только части патогенного организма, например, характерный для него белок, на распознавание которого мы и хотим настроить иммунную систему. В упомянутом в начале статьи списке ВОЗ присутствует всего 9 аттенуированных вакцин, остальные же представляют из себя различные рекомбинантные варианты.
В случае коронавируса SARS-CoV-2 таким характерным белком может служить белок SPIKE. В его составе находится домен RBD, который связывается с человеческим белком ACE2, с помощью чего генетический материал вируса попадает в здоровую клетку. Если облепить антителами SPIKE или только RBD, вирус лишится возможности оставлять свой генетический материал там, где его не просили. Более того, и этот домен, и весь белок довольно консервативны — то есть, при всей скорости изменения вируса, белок мутирует очень медленно. Значит, вакцины, основанные на этом белке, останутся эффективными в течение долгого времени.
Конечно, без ложки дегтя никуда — никто не отменял ADE-эффект. Он заключается в том, что вирусы, связанные с антителами, могут начать проникать в клетки иммунной системы человека и размножаться в них, так как будут захватываться как патогены. Чаще всего это связано со стерикой взаимодействия антитела и вируса, а также низкой аффинностью (силой связывания). Но все же, если сильно постараться, можно сделать вакцину эффективной и безопасной.
Из вышесказанного понятно, что работать следует с белком SPIKE или его частью. Но как найти этот SPIKE? Как найти и выделить из него нужный домен? И как синтезировать его для вакцины так, чтобы не перезаражать всех, кто отважился над этим работать? Вот здесь и помогут методы рационального дизайна и биоинформатики. Ниже мы приведем несколько простых примеров конкретных задач, которые можно решить с их помощью.
Итак, мы знаем, что хотим работать с белком SPIKE или его частью RBD. Получить белок можно только с помощью живых клеток, которые синтезируют его на матрице РНК. Работа с РНК — это сложно, хотя бы потому, что она очень нестабильная, но клетки могут получить ее сами с матрицы ДНК. Именно ДНК нужного белка — его ген, мы можем тем или иным образом разместить в клетках, чтобы получить — наработать искомый белок. А как получить ген?
Ген белка SPIKE можно вытащить напрямую из вируса с помощью полимеразной цепной реакции (ПЦР). Но работа с настоящим полноценным вирусом — это опасно, для этого требуется внедрять и применять особые меры безопасности сотрудников и получить лицензию на каждое помещение, где такие работы будут проводиться. Можно решить проблему иначе: собрать нужный ген с нуля.
Длина гена SPIKE — несколько тысяч нуклеотидов. Для ПЦР это ерунда, но раз мы отказались от использования вирусного генома, у нас нет «матрицы», по которой можно провести его синтез. Значит, нужно делать сборку химическим синтезом, собирая весь ген по одному нуклеотиду. И вот здесь начинаются сложности: химический синтез не потянет такую длину конструкции. Вероятность прицепить неправильный нуклеотид увеличивается при увеличении длины: получение таким методом последовательности длины N без единой ошибки можно примерно оценить как 0.985^N, так что, синтезируя всю конструкцию сразу, мы получим много ошибок. А много ошибок — это совсем не тот ген, который мы хотели. Но и здесь есть выход: можно собрать только маленькие фрагменты (по 60–80 нуклеотидов), а потом «склеить» их в полный ген с помощью все той же ПЦР. Но, конечно, без нюансов обойтись не может.
Первая проблема, с которой мы можем столкнуться, — это образование «шпильки» на коротком одноцепочечном фрагменте ДНК за счет комплементарного «залипания» одной его части на другую. Такая плотная упаковка фрагмента может неэффективно расплетаться на температурах течения ПЦР и, как итог, такой фрагмент просто выпадет из реакции, в которой уже не получится целевой ген. Другой проблемой может стать энергетическая выгода связывания несоседних фрагментов. Так может получиться продукт, из которого исчезнет целый фрагмент интересующего нас гена, поскольку «склеиваться» будут преимущественно далекие друг от друга фрагменты. Наконец, последней простой (для понимания) проблемой этого рода может стать низкая энергия комплементарного взаимодействия соседних фрагментов. Как итог, на высоких температурах ПЦР не будет происходить их объединение, и итоговый продукт будет представлять из себя несколько несвязанных кусков, а не один целевой ген.
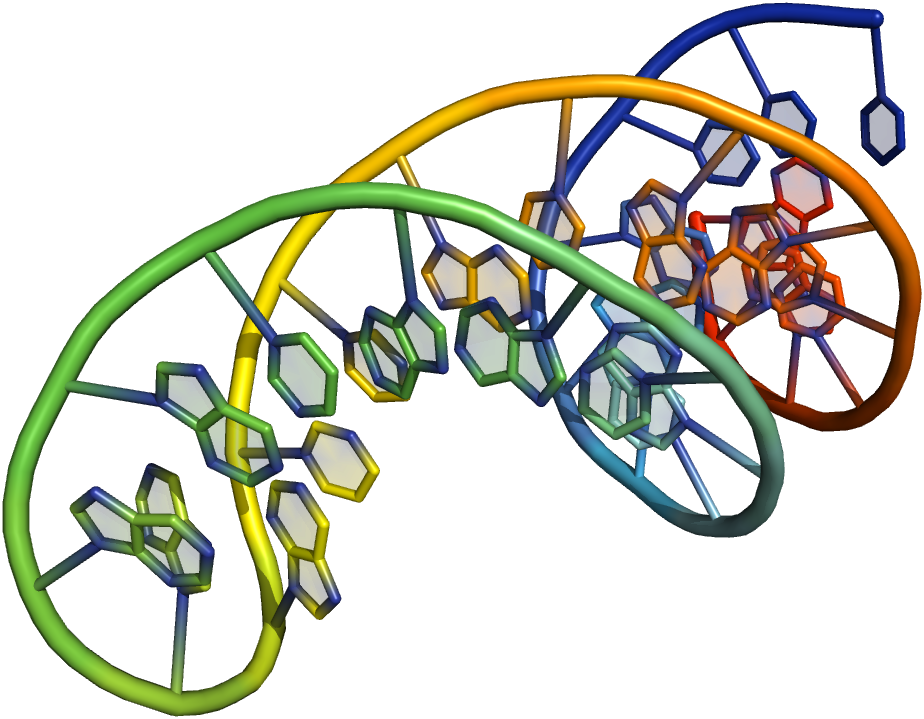
Пример плотно упакованного ДНК фрагмента.
В случае, если нам нужно четко сохранить интересующую нас нуклеотидную последовательность ДНК, у нас есть всего один управляемый параметр оптимизации — длина фрагментов, из которых осуществляется сборка. При этом целевых параметров у нас целых три. На практике мы часто можем позволить себе и изменения в самой последовательности.
Дело в том, что основная наша задача — получить белок, определяющийся последовательностью аминокислот. Протеиногенных аминокислот 20, а нуклеотидов 4. В 1961 году Фрэнсис Уотсон и Джеймс Крик постулировали, что каждый триплет нуклеотидов (кодон) кодирует какую-то из двадцати аминокислот либо специальный терминальный сигнал окончания белка — стоп-кодон. Итого, есть 3^4 = 64 способа закодировать 20 аминокислот + 1 стоп-символ. Выходит, каждую аминокислоту можно закодировать примерно тремя способами, а для получения белка длины N можно использовать порядка 3^N различных генов!
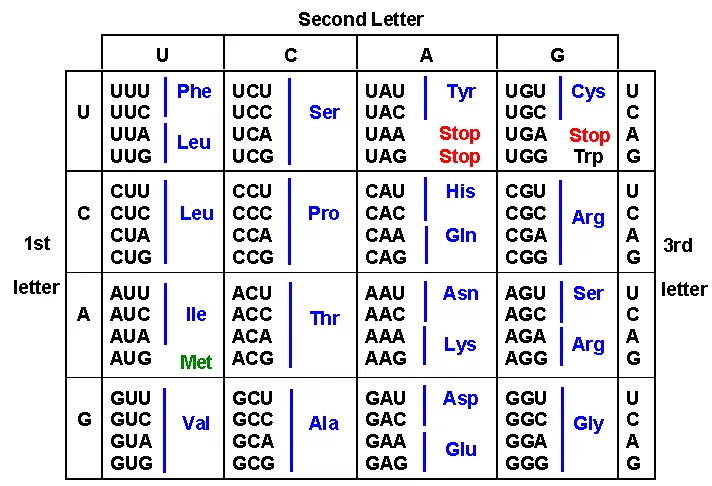
Таблица переходов от кодонов к аминокислотам.
Итак, смена кодонов на синонимичные — еще один управляемый параметр оптимизации. Но здесь же появляется и еще ряд целевых. Не все гены будут одинаково охотно использоваться клетками для синтеза белка. Эффективность процесса трансляции будет зависеть от плотности упаковки структуры транскрибированной с гена мРНК, а также от частоты встречаемости в данном целевом организме транспортной РНК, подносящей те или иные аминокислоты в зависимости от запрашиваемого кодона. Так, получив не слишком оптимальный ген, можно поставить крест на наработке нужного белка — будь то SPIKE или RBD — в больших количествах.
Здесь и возникает сразу несколько биоинформатических задач. Во-первых, было бы неплохо уметь предсказывать структуры ДНК и РНК, причем не только отдельных молекул, но и, например, парных комплексов. Над этой задачей исследователи работают уже почти 50 лет, а свежие версии алгоритмов продолжают выходить по сей день. Во-вторых, мы имеем задачу дискретной комбинаторной многопараметрической оптимизации в огромном пространстве. Мы в свое время разработали и продолжаем развивать инструмент, решающий эту задачу.
Таким образом возможно собрать любую интересующую нас генетическую конструкцию, даже очень большой длины. Но нужно ли это всегда? Выше мы уже обсуждали, что предметом нашего интереса может стать только домен RBD белка SPIKE. Поговорим о нем подробнее.
Мы знаем, что хотим научить организм облеплять вирус антителами, препятствуя его проникновению в клетки. И даже знаем, что за проникновение вируса отвечает белок SPIKE. Но этот белок — огромная махина, торчащая из вирусного капсида, и мы уже упоминали, что самая важная его часть — домен RBD, связывающийся с рецептором ACE2. Более того, едва ли будет много толку, если человеческая иммунная система будет распознавать белок SPIKE, но не будет ему мешать делать свои грязные дела (и да, мы все еще помним про ADE-эффект). Значит, давайте найдем RBD и будем фокусировать иммунную систему лишь на нем, не мешая ей лишними фрагментами SPIKE. Но это не так просто: нам необходимо, чтобы трехмерная структура отдельного RBD была точно такой же, как его структура в составе SPIKE. Иначе выработанный иммунитет к RBD просто не узнает его в новом окружении.
Вирус SARS-CoV-2. Красным отмечены тримеры белков SPIKE на его поверхности.
Понятно, что для выполнения этой задачи необходимо изучить структуру белка SPIKE. Сейчас, спустя столько времени после начала эпидемии, она уже разрешена, и не смотрел на нее только очень ленивый структурный биоинформатик. Но когда мы только начинали работы по разработке вакцины, еще никто не успел ее получить. Казалось бы, ситуация безвыходная. Но не тут-то было!
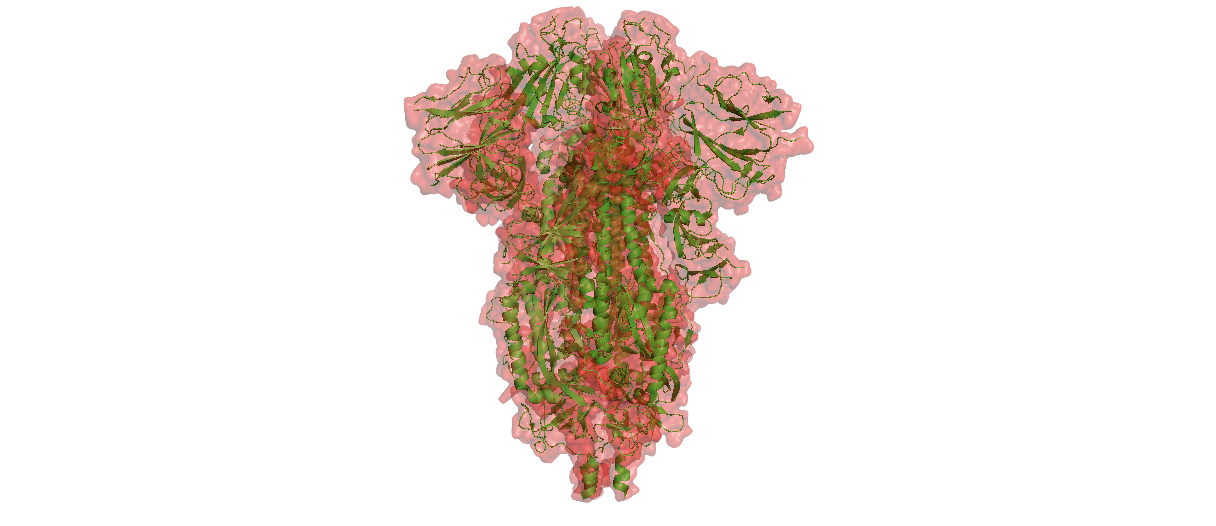
Структура изолированного тримера SPIKE из SARS-CoV-2. Нижняя часть вырастает из оболочки вируса, а верхняя торчит наружу и взаимодействует с клетками человеческого организма.
У вируса SARS-CoV-2 есть родственники, например, вирус SARS-CoV. Он активно изучался в 2000-х в связи с тем, что именно этот вирус был ответственен за вспышку атипичной пневмонии 2002–2004 годов. Он тоже относится к семейству коронавирусов, и трехмерная структура его белка SPIKE известна. Большим подарком стало и то, что родственники эти очень похожи по последовательности. А домен RBD у них вообще почти одинаковый. А это значит, что мы могли рассчитывать на то, что и структура у них будет очень похожа. Так что на основе структуры старого SPIKE мы построили структуру нужного нам нового SPIKE методом гомологичного фолдинга (подробнее про него — на курсах по структурной биоинформатике). Позднее оказалось, они действительно очень похожи, так что мы в свое время не прогадали.
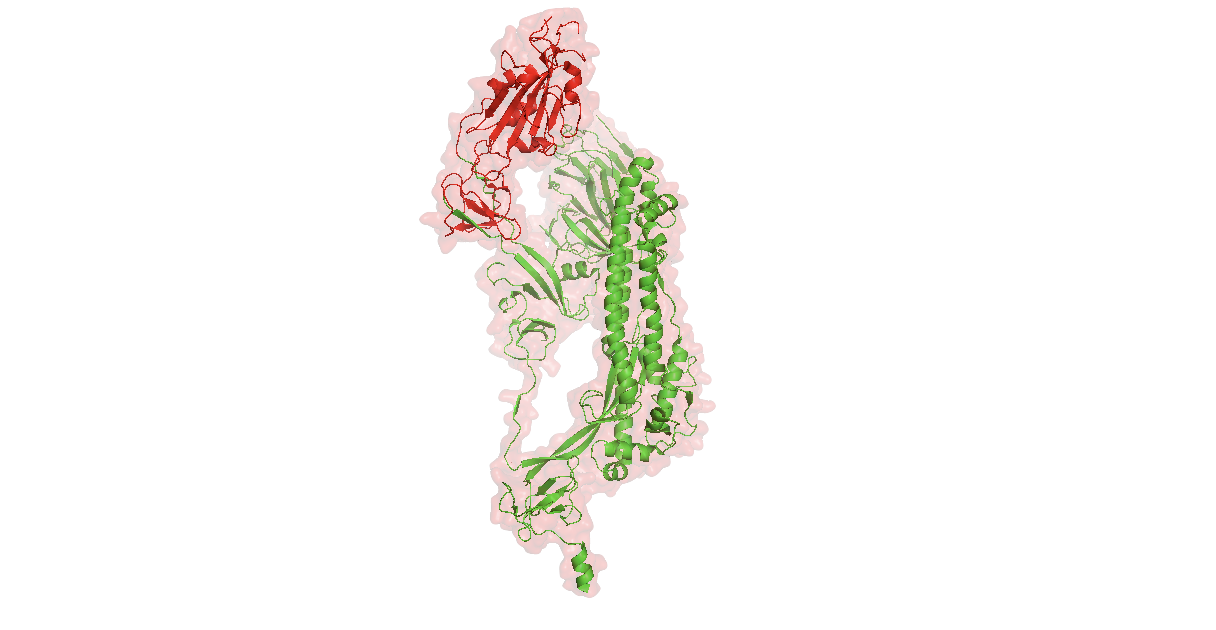
Один изолированный белок SPIKE с подсвеченным красным RBD доменом.
Итак, структура у нас есть. Теперь надо вычленить из нее RBD. Да не просто вычленить, а гарантировать, что он сохранит свою структуру отдельно от SPIKE. И хорошая тактика — выбрать несколько вариантов и потом in silico (т. е. программно, без проведения «мокрого» эксперимента в лаборатории) проверить их стабильность. Варианты не стоит подбирать путем перебора — наличие структуры позволяет нам вычислить, какие ковалентные, гидрофобные и электростатические взаимодействия стабилизируют структуру домена. Ориентируясь на их сохранение, можно подобрать потенциально очень стабильные кандидаты.
До передачи в лабораторию проверить их стабильность можно с помощью поиска минимума энергии и моделирования методом молекулярной динамики. Для этого нужно правильно их параметризовать — подобрать математические уравнения и их параметры, которые правильно и физично опишут поведение этих доменов во времени. Вообще это довольно длительные расчеты, но если у вас внезапно есть хороший вычислительный кластер с большим количеством GPU, то сделать это очень просто и быстро. У нас такой есть. Так что после всех ухищрений мы знаем, какая последовательность RBD пойдет в наши вакцины.
Увы, не во все вакцины можно запихнуть такой большой домен. RBD хоть и не SPIKE, но все равно великоват — больше 250 аминокислот, тогда как в некоторые вектора (часть вакцины, являющаяся «носителем» презентуемого белка) допустимо использовать белки максимум в 150 аминокислот, и нужно среди многообразия таких последовательностей в вирусе выбрать наиболее иммуногенные.
Выше мы уже упоминали про T-клеточный иммунитет и то, что белки MHC II на антиген-презентирующих клетках могут показать иммунной системе небольшие пептиды, стимулируя ее работу. И чтобы простимулировать работу иммунной системы максимально эффективно, необходимо знать, какой пептид в паре с каким MHC II будет наиболее иммуногенным. В таком случае мы сможем использовать очень простую идею в построении вакцины: вместо целого белка мы покажем организму несколько иммуногенных пептидов (чтоб наверняка), соединенных разлагаемыми линкерами. Антиген-презентирующие клетки с помощью белков-протеаз разрежут такие конструкции на отдельные пептиды и представят Т-лимфоцитам. Выглядит классно, но, как обычно, не все так просто.
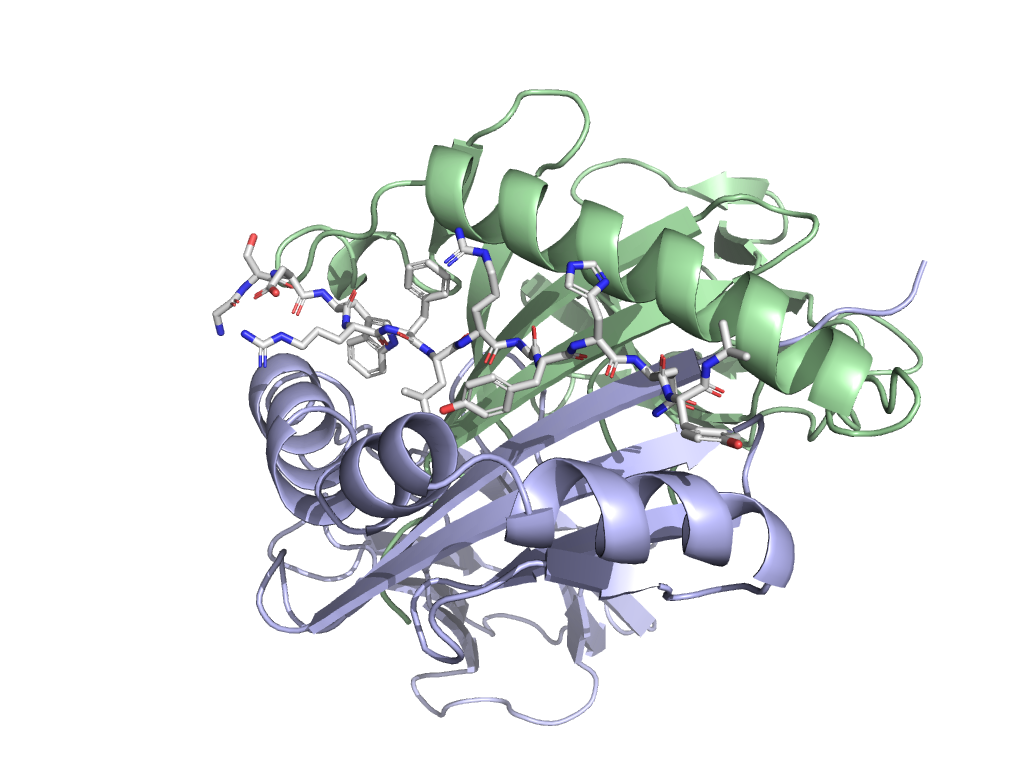
MHC II класса в комплексе с пептидом VGSDWRFLRGYHQYA
Проблема в том, что коротких пептидов из большого белка получить можно много (
И вот у нас есть набор иммуногенных пептидов, и появляется следующая задача: соединить эти пептиды между собой так, чтобы протеазы, которые разрезают белки на пептиды, разрезали нашу полипептидную цепочку именно там, где надо. Ведь если разрез будет происходить в случайном месте, MHC II класса покажут иммунитету не то, что мы хотели, а «химерные» пептиды, не имеющие отношения к SARS-CoV-2. Так как протеаз много разных (есть ощущение, что все мы до сих пор не знаем), а данных по ним и их излюбленным местам разреза — еще больше, для решения этой задачи мы применяем аналитику и машинное обучение.
Разработка препарата — это не только подбор последовательности, которая будет действовать каким-либо образом. Это понимание сути подобного действия в живых многоклеточных организмах, особенно в людях.
Для этого нужно знать, сколько препарат сохраняется в организме и как его концентрация связана с оказываемым терапевтическим эффектом. Разумеется, и то и другое можно померить. Но замеры — например, взятие крови или, еще хуже, биопсии, — это не всегда простая задача. Так, мышь после взятия образца крови чаще всего погибает, а уничтожать тысячи мышей за исследование мы не можем. Пациенты, участвующие в клиническом исследовании, не хотят, чтобы у них брали кровь каждые пятнадцать минут, и их можно понять.
Что это означает? Это означает, что точек мало. При этом исходные дозы исследуются далеко не в полном объеме, и требуется составлять математические модели, которые бы показывали фармакокинетику — концентрацию препарата от времени, дозы и характеристик организма — и фармакодинамику — действие текущей концентрации препарата на целевые показатели. Модели эти зачастую представляют из себя систему дифференциальных уравнений, решения которой способны делать хорошие приближения и обладают прогностической силой.
Рассмотрим пример. Предположим, у нас есть препарат в виде таблетки, пациент ее принимает, и через некоторое время активное вещество таблетки попадает в кровоток. Когда мы берем у человека кровь, мы можем определить значения концентрации активного вещества и получить точки во времени. Выглядит это как-то так:
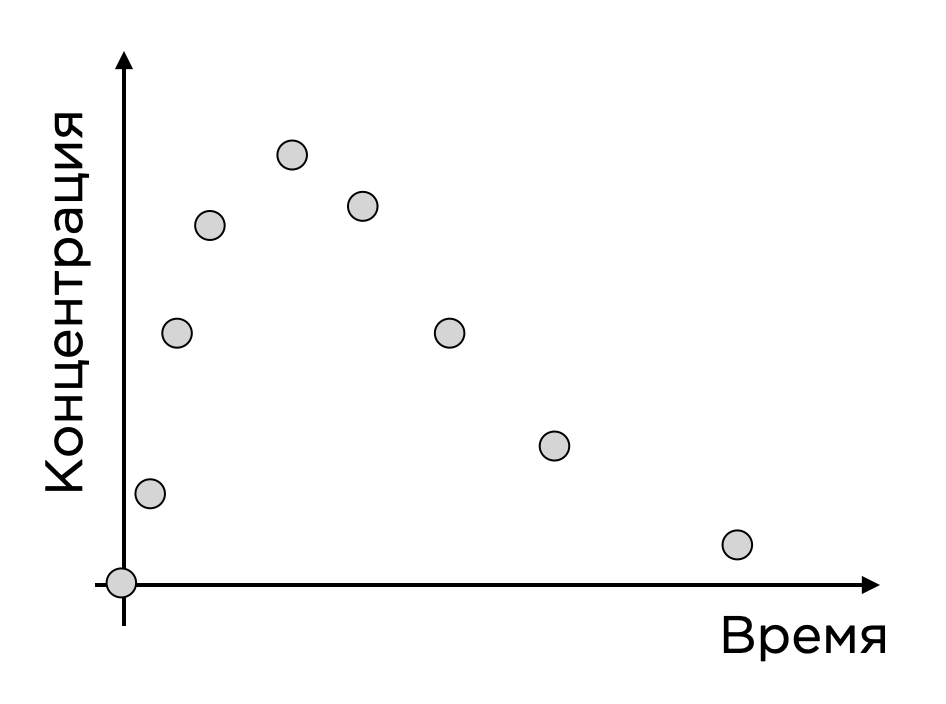
Такая точечная функция весьма ограничена, и у нас нет представления о том, что будет происходить в любой момент времени и, что еще важнее, при других дозовых уровнях (ведь мы могли выбрать в исследование что-то неоптимальное), а хотелось бы это представление иметь.
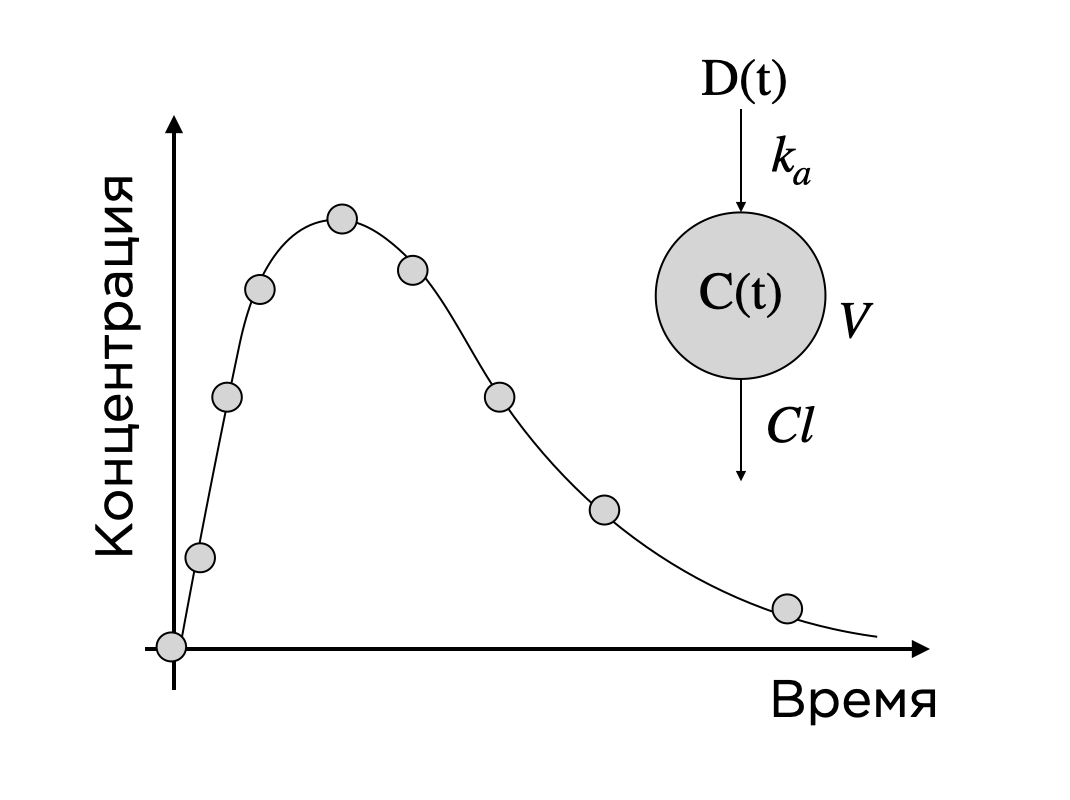
Тогда мы строим зависимость, исходя из простого предположения: препарат — фиксированная доза — попадает в организм, всасывается с некоторой скоростью ka, далее распространяется по кровотоку объема V (что мы и видим на графике время-концентрация) и с некоторой скоростью Cl из организма выводится. Изменение концентрации во времени — это производная концентрации по времени. Приправим этот факт щепоткой закона действующих масс, и вот уже вырисовываются дифференциальные уравнения. Решение системы, подчиняющейся этим законам и приближенное к полученным точкам, будет отражать реальную зависимость концентрации от времени, то есть фармакокинетику (ФК) препарата. При этом, меняя начальное условие — дозу вводимого препарата, мы можем показать, какая ФК будет в этом случае — ведь параметры ka, V и Cl отражают процессы и объекты, предположительно не зависимые от дозы. Разумеется, в реальных задачах такие модели могут выглядеть гораздо сложнее, учитывать циркуляцию препарата по тканям, взаимодействие различных препаратов друг с другом, популяционные особенности организма тех или иных людей (например, с почечной недостаточностью, мешающей выводу препарата из организма) и так далее.
Аналогичным образом можно поступить и с эффектом. Опять рассмотрим пример, когда наш препарат влияет на синтез какого-либо белка. Сходным образом рисуем схему, записываем уравнения с учетом влияния концентрации и приближаем решение к данным.
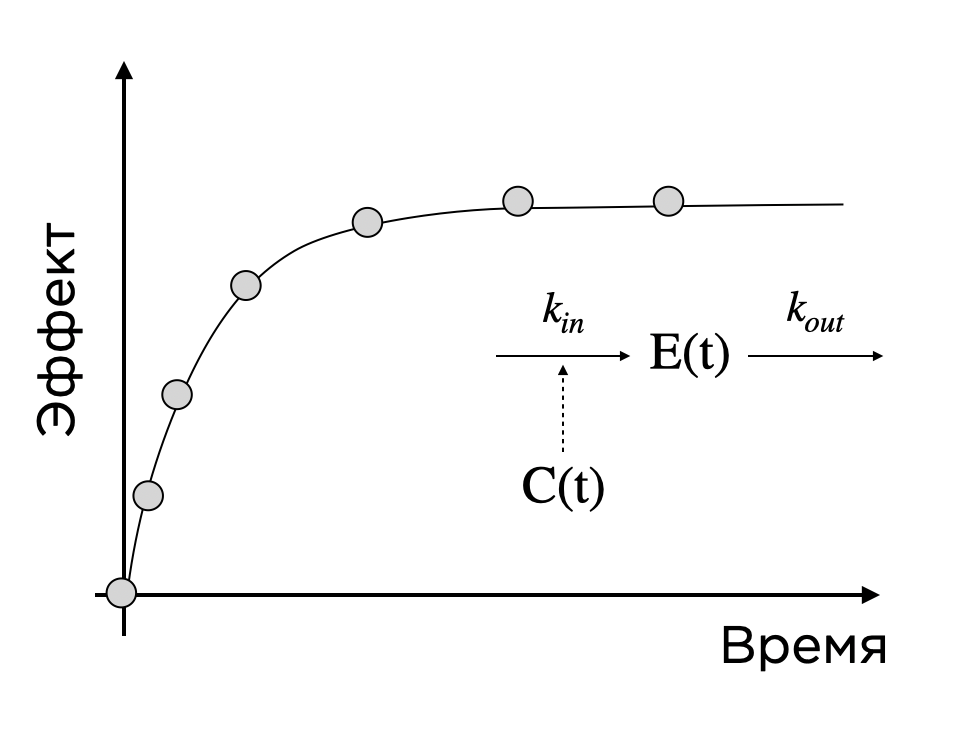
Таким образом, у нас есть зависимость концентрации от времени и дозы (если мы меняем начальное условие) C(t) и зависимость эффекта от концентрации и времени E(t). Соединяя эти сущности, получаем ФКФД-модель, которая, к примеру, может показать, какая доза будет давать тот или иной эффект. И все это — на вашем компьютере! Вновь это лишь верхушка айсберга: сложные биологические системы обычно описываются целой цепочкой взаимосвязанных процессов, а используемые препараты часто влияют сразу на несколько компонентов таких систем.
Внимательный читатель мог заметить, что всем вышесказанном речь шла не о ФК/ФД-моделировании для вакцин. Почему? В случае с вакциной не очень понятно, что считать за ФК и как измерять ФД. Однако исследователи не сдаются, и математическое моделирование заявляет свои права и на эту область.
На простых для восприятия примерах мы рассказали о некоторых задачах, которые биоинформатики выполняют очень быстро, если на пороге вирус, захватывающий весь мир. Однако в действительности эти и многие другие задачи мы решаем каждый день. Никакая пандемия не отменяет того, что люди страдают от онкологических и аутоиммунных заболеваний, а дети рождаются с тяжелейшими генетическими дефектами. Создание препаратов, способных помочь таким пациентам, ничуть не менее важно, а возникающие при этом задачи обычно даже сложнее и требуют гораздо больше времени на проведение исследований.
Для создания терапии при таких болезнях мы разрабатываем как биологические, так и низкомолекулярные соединения. Первые требуют генетические конструкции, оптимизацию работы живых клеток и определение сложных физико-химических характеристик получающихся белков и других биологических молекул (например, мы делаем терапевтические вирусы для лечения орфанных заболеваний). Для вторых появляются проблемы течения органического синтеза, проницаемости клеточных мембран, стабильности в различных фракциях микросом и так далее. Чтобы создать такие большие и малые молекулы, мы применяем методы рационального дизайна на основе трехмерных полноатомных моделей молекул и их мишеней, рассчитываем способы связывания терапевтических молекул с их мишенями, оптимизируем схемы химического синтеза, предсказываем зависимость поведения препарата в организме в зависимости от роста и веса пациента и многое другое.
Разумеется, для создания лекарственных средств нужны высококлассные специалисты, как в биологических и химических «мокрых» лабораториях, так и в «сухих» областях вычислительной биологии и биоинформатики. В России нет вузов, которые выпускают готовых вычислительных биологов, ориентированных на решение задач современной разработки лекарственных средств. Долгое время мы обучали специалистов внутри BIOCAD и вырастили не одно поколение востребованных специалистов. Теперь же пришло время открыть полноценную программу по обучению биоинформатике, и преподаваться она будет магистрантам Питерской Вышки. Старт программы — сентябрь 2021 года, но уже в 2020 мы запускаем пилотный трек в рамках уже имеющейся программы «Программирование и анализ данных».
Мы очень ждем студентов с сильным физико-математическим бэкграундом, без химической и биологической подготовки. Преподаватели Питерской Вышки обеспечат лучшие в стране курсы по алгоритмам, программированию, анализу данных, а сотрудники индустрии расскажут спецглавы физики, молекулярной биологии, химии, а также специальные курсы по молекулярным симуляциям, алгоритмам структурной биоинформатики, системной фармакологии и иным важным для области темам. И, что тоже важно, расскажем о настоящих индустриальных биологических задачах и научим использовать полученные знания и навыки для их решения.
Скоро информация о новой программе появится на сайте Санкт-петербургской школы физико-математических и компьютерных наук. Следите за новостями!
В СМИ активно раскручиваются обсуждения клинических исследований, производства и будущего применения вакцин, тогда как «ранние» разработчики, скорее, отмахивались от излишней публичности. Теперь же вся часть по дизайну и сборке кандидатов завершена, мы затаились в ожидании результатов с животных моделей и получения разрешений на людях. Так что можно выделить немного времени для того, чтоб рассказать какие задачи возникают внутри.
Поскольку Habr — ресурс в первую очередь околоайтишный, а в современной фармацевтике активно применяются методы биоинформатики и вычислительной биологии, мы остановимся именно на «сухих» математических задачах. На самом деле эти задачи выбраны еще и потому, что в следующем году НИУ ВШЭ — Санкт-Петербург и BIOCAD открывают свою магистратуру по вычислительной биологии, но об этом в самом конце. В первую же очередь разберемся, что, собственно, нужно было сделать.
Немного об авторах
Над этой статьей работали три сотрудника BIOCAD:
Диана Кондинская — выпускница биофизики физического факультета СПбГУ, старший специалист компании по структурной биоинформатике. Своими руками делает белковые дизайны оригинальных препаратов, занимается молекулярными симуляциями и всем, что с ними связано.
Александра Черткова — выпускница биоинформатики СПбГПУ. Занимает должность руководителя отдела системной биологии, отвечает за моделирование фармакокинетики и фармакодинамики всех доклинических и клинических исследований компании.
Павел Яковлев окончил программу по биоинформатике в Академическом Университете. В ходе выполнения первого же научно-исследовательского проекта в университете попал в BIOCAD, оказавшись там первым биоинформатиком. Последовательно прошел должности руководителя группы биоинформатики и директора департамента вычислительной биологии. С февраля 2020 года стал руководить всеми исследованиями в области разработки оригинальных препаратов на должности директора по ранней разработке и развитию.
Что такое вакцины
Говоря простым языком, вакцины — это тип лекарственных препаратов, задача которых — «научить» иммунную систему находить и уничтожать определенные патогены. Традиционно при этом подразумеваются профилактические вакцины, которые тренируют иммунитет еще до попадания патогена в организм, но в некоторых областях бывают и исключения, например, терапевтические онковакцины.
Рассказывать в двух словах про работу иммунной системы высших млекопитающих — дело неблагодарное. Курс иммунологии на любом биофаке традиционно считается одним из самых сложных, да еще и устаревает обычно быстрее, чем идет семестр. Так что, ни в коем случае не претендуя на полноту, обратимся только к главным интересующим нас деталям адаптивного иммунитета, вырабатывающегося организмом индивидуально к новым угрозам.
Первым важным элементом является В-клеточный иммунитет. Его суть заключается в постоянном (на протяжении всей жизни!) образовании новых клеток, называемых В-лимфоцитами. Каждый такой лимфоцит способен распознавать уникальную мишень с помощью своего В-клеточного рецептора. Также эти клетки способны производить специальную форму этих рецепторов — иммуноглобулины (или антитела), которые не остаются на поверхности клетки, а плавают свободно, специфически связывая патогенные белки как в свободной форме, так и на поверхности клеток. Такие антитела способны не только заблокировать какое-либо взаимодействие, но и пометить клетку на уничтожение другими компонентами иммунной системы — фагоцитами и натуральными киллерами. Кстати, синтетические антитела являются основой для современной противораковой терапии.
Наличие В-клеточного иммунного ответа подразумевает некоторую встроенную в организм систему «свой-чужой», которая бы позволила идентифицировать внешние патогены и критичные мутации собственных белков и вызвать на них иммунную реакцию. Действительно, абсолютно каждая клетка нашего организма содержит систему, обеспечивающую презентацию производимых клеткой белков на поверхности. Так, специальный белковый комплекс, называемый протеасомой, способен расщеплять белки внутри клетки на короткие линейные фрагменты — пептиды, состоящие не более чем из пары десятков аминокислот. Такие пептиды могут связываться с продуктами аллелей HLA, кодирующих специальные белки MHC (в данном случае I типа). Несмотря на то, что этих аллелей в человеческом геноме заложено несколько тысяч (см., например, здесь), активно работают всего несколько сотен, при этом в каждом человеке свои. Это определяется как наследственностью, так и внешними факторами, так что обычно справедливо предполагать, что у жителей одного региона большая часть активных MHC будет одинаковой. Связываясь с продуктами протеасомной деградации, МНС I типа выходят на мембрану клетки, таким образом показывая продукты, вырабатываемые клеткой.
Помимо механизма презентации своих внутренностей каждой клеткой, существуют также профессиональные демонстраторы — антиген-презентирующие клетки. Они способны захватывать объекты из внешней среды и показывать их пептиды аналогичным образом, используя для этого уже MHC II класса, которые также являются продуктами HLA.
Но раз кто-то демонстрирует продукты, какая-то система должна их и распознавать. За это отвечает вторая важная ветвь адаптивной иммунной системы — Т-клеточный иммунитет. С помощью специальных рецепторов (называемых, очевидно, Т-клеточными) такие клетки способны распознавать пептиды, не соответствующие нормальным белкам организма и вызывать различные иммунные реакции. Клетки T-хелперы будут смотреть на комплексы из MHC II класса и презентируемых ими пептидов и активировать В-клеточный иммунитет и ряд других реакций. Цитотоксические клетки Т-киллеры просто будут уничтожать всех, чья пара из MHC I класса и презентуемого пептида им не понравится.
Вооружившись этим знанием, теперь мы можем осознать истинную задачу вакцины — обеспечить в организме наличие белков патогена (в нашем случае вируса SARS-CoV-2), чтобы запустить выработку В- и/или Т-клеточного иммунитета, который будет способен быстро нейтрализовать сам патоген при его попадании в организм. Вырабатываемые антитела будут препятствовать проникновению вируса в клетку, а Т-лимфоциты смогут убивать клетки, в которых вирус уже поселился.
Очень часто в качестве вакцин используют ослабленные патогенные микроорганизмы — вирусы и бактерии, которые несут на себе характерные распознаваемые иммунной системой белки, но сами по себе безвредны. Такие вакцины называются аттенуированными. Тем не менее, разработка и доказательство их безопасности — сложный и трудоемкий процесс. В качестве эффективной безопасной альтернативы можно использовать рекомбинантные вакцины, созданные с помощью генно-инженерных методов. В рекомбинантных вакцинах используются только части патогенного организма, например, характерный для него белок, на распознавание которого мы и хотим настроить иммунную систему. В упомянутом в начале статьи списке ВОЗ присутствует всего 9 аттенуированных вакцин, остальные же представляют из себя различные рекомбинантные варианты.
В случае коронавируса SARS-CoV-2 таким характерным белком может служить белок SPIKE. В его составе находится домен RBD, который связывается с человеческим белком ACE2, с помощью чего генетический материал вируса попадает в здоровую клетку. Если облепить антителами SPIKE или только RBD, вирус лишится возможности оставлять свой генетический материал там, где его не просили. Более того, и этот домен, и весь белок довольно консервативны — то есть, при всей скорости изменения вируса, белок мутирует очень медленно. Значит, вакцины, основанные на этом белке, останутся эффективными в течение долгого времени.
Конечно, без ложки дегтя никуда — никто не отменял ADE-эффект. Он заключается в том, что вирусы, связанные с антителами, могут начать проникать в клетки иммунной системы человека и размножаться в них, так как будут захватываться как патогены. Чаще всего это связано со стерикой взаимодействия антитела и вируса, а также низкой аффинностью (силой связывания). Но все же, если сильно постараться, можно сделать вакцину эффективной и безопасной.
Из вышесказанного понятно, что работать следует с белком SPIKE или его частью. Но как найти этот SPIKE? Как найти и выделить из него нужный домен? И как синтезировать его для вакцины так, чтобы не перезаражать всех, кто отважился над этим работать? Вот здесь и помогут методы рационального дизайна и биоинформатики. Ниже мы приведем несколько простых примеров конкретных задач, которые можно решить с их помощью.
Задача 1: получить генетические конструкции
Итак, мы знаем, что хотим работать с белком SPIKE или его частью RBD. Получить белок можно только с помощью живых клеток, которые синтезируют его на матрице РНК. Работа с РНК — это сложно, хотя бы потому, что она очень нестабильная, но клетки могут получить ее сами с матрицы ДНК. Именно ДНК нужного белка — его ген, мы можем тем или иным образом разместить в клетках, чтобы получить — наработать искомый белок. А как получить ген?
Ген белка SPIKE можно вытащить напрямую из вируса с помощью полимеразной цепной реакции (ПЦР). Но работа с настоящим полноценным вирусом — это опасно, для этого требуется внедрять и применять особые меры безопасности сотрудников и получить лицензию на каждое помещение, где такие работы будут проводиться. Можно решить проблему иначе: собрать нужный ген с нуля.
Если вы запамятовали, как работает PCR, этот ролик позволит быстро освежить воспоминания
Длина гена SPIKE — несколько тысяч нуклеотидов. Для ПЦР это ерунда, но раз мы отказались от использования вирусного генома, у нас нет «матрицы», по которой можно провести его синтез. Значит, нужно делать сборку химическим синтезом, собирая весь ген по одному нуклеотиду. И вот здесь начинаются сложности: химический синтез не потянет такую длину конструкции. Вероятность прицепить неправильный нуклеотид увеличивается при увеличении длины: получение таким методом последовательности длины N без единой ошибки можно примерно оценить как 0.985^N, так что, синтезируя всю конструкцию сразу, мы получим много ошибок. А много ошибок — это совсем не тот ген, который мы хотели. Но и здесь есть выход: можно собрать только маленькие фрагменты (по 60–80 нуклеотидов), а потом «склеить» их в полный ген с помощью все той же ПЦР. Но, конечно, без нюансов обойтись не может.
Первая проблема, с которой мы можем столкнуться, — это образование «шпильки» на коротком одноцепочечном фрагменте ДНК за счет комплементарного «залипания» одной его части на другую. Такая плотная упаковка фрагмента может неэффективно расплетаться на температурах течения ПЦР и, как итог, такой фрагмент просто выпадет из реакции, в которой уже не получится целевой ген. Другой проблемой может стать энергетическая выгода связывания несоседних фрагментов. Так может получиться продукт, из которого исчезнет целый фрагмент интересующего нас гена, поскольку «склеиваться» будут преимущественно далекие друг от друга фрагменты. Наконец, последней простой (для понимания) проблемой этого рода может стать низкая энергия комплементарного взаимодействия соседних фрагментов. Как итог, на высоких температурах ПЦР не будет происходить их объединение, и итоговый продукт будет представлять из себя несколько несвязанных кусков, а не один целевой ген.
Пример плотно упакованного ДНК фрагмента.
В случае, если нам нужно четко сохранить интересующую нас нуклеотидную последовательность ДНК, у нас есть всего один управляемый параметр оптимизации — длина фрагментов, из которых осуществляется сборка. При этом целевых параметров у нас целых три. На практике мы часто можем позволить себе и изменения в самой последовательности.
Дело в том, что основная наша задача — получить белок, определяющийся последовательностью аминокислот. Протеиногенных аминокислот 20, а нуклеотидов 4. В 1961 году Фрэнсис Уотсон и Джеймс Крик постулировали, что каждый триплет нуклеотидов (кодон) кодирует какую-то из двадцати аминокислот либо специальный терминальный сигнал окончания белка — стоп-кодон. Итого, есть 3^4 = 64 способа закодировать 20 аминокислот + 1 стоп-символ. Выходит, каждую аминокислоту можно закодировать примерно тремя способами, а для получения белка длины N можно использовать порядка 3^N различных генов!
Таблица переходов от кодонов к аминокислотам.
Итак, смена кодонов на синонимичные — еще один управляемый параметр оптимизации. Но здесь же появляется и еще ряд целевых. Не все гены будут одинаково охотно использоваться клетками для синтеза белка. Эффективность процесса трансляции будет зависеть от плотности упаковки структуры транскрибированной с гена мРНК, а также от частоты встречаемости в данном целевом организме транспортной РНК, подносящей те или иные аминокислоты в зависимости от запрашиваемого кодона. Так, получив не слишком оптимальный ген, можно поставить крест на наработке нужного белка — будь то SPIKE или RBD — в больших количествах.
Здесь и возникает сразу несколько биоинформатических задач. Во-первых, было бы неплохо уметь предсказывать структуры ДНК и РНК, причем не только отдельных молекул, но и, например, парных комплексов. Над этой задачей исследователи работают уже почти 50 лет, а свежие версии алгоритмов продолжают выходить по сей день. Во-вторых, мы имеем задачу дискретной комбинаторной многопараметрической оптимизации в огромном пространстве. Мы в свое время разработали и продолжаем развивать инструмент, решающий эту задачу.
Таким образом возможно собрать любую интересующую нас генетическую конструкцию, даже очень большой длины. Но нужно ли это всегда? Выше мы уже обсуждали, что предметом нашего интереса может стать только домен RBD белка SPIKE. Поговорим о нем подробнее.
Задача 2: найди RBD
Мы знаем, что хотим научить организм облеплять вирус антителами, препятствуя его проникновению в клетки. И даже знаем, что за проникновение вируса отвечает белок SPIKE. Но этот белок — огромная махина, торчащая из вирусного капсида, и мы уже упоминали, что самая важная его часть — домен RBD, связывающийся с рецептором ACE2. Более того, едва ли будет много толку, если человеческая иммунная система будет распознавать белок SPIKE, но не будет ему мешать делать свои грязные дела (и да, мы все еще помним про ADE-эффект). Значит, давайте найдем RBD и будем фокусировать иммунную систему лишь на нем, не мешая ей лишними фрагментами SPIKE. Но это не так просто: нам необходимо, чтобы трехмерная структура отдельного RBD была точно такой же, как его структура в составе SPIKE. Иначе выработанный иммунитет к RBD просто не узнает его в новом окружении.
Вирус SARS-CoV-2. Красным отмечены тримеры белков SPIKE на его поверхности.
Понятно, что для выполнения этой задачи необходимо изучить структуру белка SPIKE. Сейчас, спустя столько времени после начала эпидемии, она уже разрешена, и не смотрел на нее только очень ленивый структурный биоинформатик. Но когда мы только начинали работы по разработке вакцины, еще никто не успел ее получить. Казалось бы, ситуация безвыходная. Но не тут-то было!
Структура изолированного тримера SPIKE из SARS-CoV-2. Нижняя часть вырастает из оболочки вируса, а верхняя торчит наружу и взаимодействует с клетками человеческого организма.
У вируса SARS-CoV-2 есть родственники, например, вирус SARS-CoV. Он активно изучался в 2000-х в связи с тем, что именно этот вирус был ответственен за вспышку атипичной пневмонии 2002–2004 годов. Он тоже относится к семейству коронавирусов, и трехмерная структура его белка SPIKE известна. Большим подарком стало и то, что родственники эти очень похожи по последовательности. А домен RBD у них вообще почти одинаковый. А это значит, что мы могли рассчитывать на то, что и структура у них будет очень похожа. Так что на основе структуры старого SPIKE мы построили структуру нужного нам нового SPIKE методом гомологичного фолдинга (подробнее про него — на курсах по структурной биоинформатике). Позднее оказалось, они действительно очень похожи, так что мы в свое время не прогадали.
Один изолированный белок SPIKE с подсвеченным красным RBD доменом.
Итак, структура у нас есть. Теперь надо вычленить из нее RBD. Да не просто вычленить, а гарантировать, что он сохранит свою структуру отдельно от SPIKE. И хорошая тактика — выбрать несколько вариантов и потом in silico (т. е. программно, без проведения «мокрого» эксперимента в лаборатории) проверить их стабильность. Варианты не стоит подбирать путем перебора — наличие структуры позволяет нам вычислить, какие ковалентные, гидрофобные и электростатические взаимодействия стабилизируют структуру домена. Ориентируясь на их сохранение, можно подобрать потенциально очень стабильные кандидаты.
До передачи в лабораторию проверить их стабильность можно с помощью поиска минимума энергии и моделирования методом молекулярной динамики. Для этого нужно правильно их параметризовать — подобрать математические уравнения и их параметры, которые правильно и физично опишут поведение этих доменов во времени. Вообще это довольно длительные расчеты, но если у вас внезапно есть хороший вычислительный кластер с большим количеством GPU, то сделать это очень просто и быстро. У нас такой есть. Так что после всех ухищрений мы знаем, какая последовательность RBD пойдет в наши вакцины.
Задача 3: иммуногенные фрагменты
Увы, не во все вакцины можно запихнуть такой большой домен. RBD хоть и не SPIKE, но все равно великоват — больше 250 аминокислот, тогда как в некоторые вектора (часть вакцины, являющаяся «носителем» презентуемого белка) допустимо использовать белки максимум в 150 аминокислот, и нужно среди многообразия таких последовательностей в вирусе выбрать наиболее иммуногенные.
Выше мы уже упоминали про T-клеточный иммунитет и то, что белки MHC II на антиген-презентирующих клетках могут показать иммунной системе небольшие пептиды, стимулируя ее работу. И чтобы простимулировать работу иммунной системы максимально эффективно, необходимо знать, какой пептид в паре с каким MHC II будет наиболее иммуногенным. В таком случае мы сможем использовать очень простую идею в построении вакцины: вместо целого белка мы покажем организму несколько иммуногенных пептидов (чтоб наверняка), соединенных разлагаемыми линкерами. Антиген-презентирующие клетки с помощью белков-протеаз разрежут такие конструкции на отдельные пептиды и представят Т-лимфоцитам. Выглядит классно, но, как обычно, не все так просто.
MHC II класса в комплексе с пептидом VGSDWRFLRGYHQYA
Проблема в том, что коротких пептидов из большого белка получить можно много (
N — K + 1, где N — длина белка, а K — длина пептида, которая может варьироваться в диапазоне 9–15 аминокислот). А варианты MHC II класса, как мы писали выше, вообще разные от популяции к популяции, от расы к расе. И одна из задач, которые приходится решать биоинформатикам, — как раз поиск таких пептидов, презентация которых на поверхности клеток с помощью MHC II вызовет хороший иммунный ответ в данной популяции. Конечно, лучшим методом был бы выбор ряда характерных MHC молекул и анализ всех возможных пептидов на связывание с этими молекулами с помощью предсказания межмолекулярных взаимодействий. Конечно, это дешевле, чем проводить подобные исследования in vitro (в «мокрой» лаборатории), но все еще дороговато. В связи с этим в этой области активно развиваются методы машинного обучения, которые по последовательности пептида предсказывают, с какими МНС он может связаться. Точность этих методов все еще оставляет желать лучшего и не позволяют получать результаты для всех случаев, но в совокупности с априорными биологическими знаниями, для каждого конкретного случая решить задачу обычно все же удается.И вот у нас есть набор иммуногенных пептидов, и появляется следующая задача: соединить эти пептиды между собой так, чтобы протеазы, которые разрезают белки на пептиды, разрезали нашу полипептидную цепочку именно там, где надо. Ведь если разрез будет происходить в случайном месте, MHC II класса покажут иммунитету не то, что мы хотели, а «химерные» пептиды, не имеющие отношения к SARS-CoV-2. Так как протеаз много разных (есть ощущение, что все мы до сих пор не знаем), а данных по ним и их излюбленным местам разреза — еще больше, для решения этой задачи мы применяем аналитику и машинное обучение.
А дальше
Разработка препарата — это не только подбор последовательности, которая будет действовать каким-либо образом. Это понимание сути подобного действия в живых многоклеточных организмах, особенно в людях.
Для этого нужно знать, сколько препарат сохраняется в организме и как его концентрация связана с оказываемым терапевтическим эффектом. Разумеется, и то и другое можно померить. Но замеры — например, взятие крови или, еще хуже, биопсии, — это не всегда простая задача. Так, мышь после взятия образца крови чаще всего погибает, а уничтожать тысячи мышей за исследование мы не можем. Пациенты, участвующие в клиническом исследовании, не хотят, чтобы у них брали кровь каждые пятнадцать минут, и их можно понять.
Что это означает? Это означает, что точек мало. При этом исходные дозы исследуются далеко не в полном объеме, и требуется составлять математические модели, которые бы показывали фармакокинетику — концентрацию препарата от времени, дозы и характеристик организма — и фармакодинамику — действие текущей концентрации препарата на целевые показатели. Модели эти зачастую представляют из себя систему дифференциальных уравнений, решения которой способны делать хорошие приближения и обладают прогностической силой.
Рассмотрим пример. Предположим, у нас есть препарат в виде таблетки, пациент ее принимает, и через некоторое время активное вещество таблетки попадает в кровоток. Когда мы берем у человека кровь, мы можем определить значения концентрации активного вещества и получить точки во времени. Выглядит это как-то так:
Такая точечная функция весьма ограничена, и у нас нет представления о том, что будет происходить в любой момент времени и, что еще важнее, при других дозовых уровнях (ведь мы могли выбрать в исследование что-то неоптимальное), а хотелось бы это представление иметь.
Тогда мы строим зависимость, исходя из простого предположения: препарат — фиксированная доза — попадает в организм, всасывается с некоторой скоростью ka, далее распространяется по кровотоку объема V (что мы и видим на графике время-концентрация) и с некоторой скоростью Cl из организма выводится. Изменение концентрации во времени — это производная концентрации по времени. Приправим этот факт щепоткой закона действующих масс, и вот уже вырисовываются дифференциальные уравнения. Решение системы, подчиняющейся этим законам и приближенное к полученным точкам, будет отражать реальную зависимость концентрации от времени, то есть фармакокинетику (ФК) препарата. При этом, меняя начальное условие — дозу вводимого препарата, мы можем показать, какая ФК будет в этом случае — ведь параметры ka, V и Cl отражают процессы и объекты, предположительно не зависимые от дозы. Разумеется, в реальных задачах такие модели могут выглядеть гораздо сложнее, учитывать циркуляцию препарата по тканям, взаимодействие различных препаратов друг с другом, популяционные особенности организма тех или иных людей (например, с почечной недостаточностью, мешающей выводу препарата из организма) и так далее.
Аналогичным образом можно поступить и с эффектом. Опять рассмотрим пример, когда наш препарат влияет на синтез какого-либо белка. Сходным образом рисуем схему, записываем уравнения с учетом влияния концентрации и приближаем решение к данным.
Таким образом, у нас есть зависимость концентрации от времени и дозы (если мы меняем начальное условие) C(t) и зависимость эффекта от концентрации и времени E(t). Соединяя эти сущности, получаем ФКФД-модель, которая, к примеру, может показать, какая доза будет давать тот или иной эффект. И все это — на вашем компьютере! Вновь это лишь верхушка айсберга: сложные биологические системы обычно описываются целой цепочкой взаимосвязанных процессов, а используемые препараты часто влияют сразу на несколько компонентов таких систем.
Внимательный читатель мог заметить, что всем вышесказанном речь шла не о ФК/ФД-моделировании для вакцин. Почему? В случае с вакциной не очень понятно, что считать за ФК и как измерять ФД. Однако исследователи не сдаются, и математическое моделирование заявляет свои права и на эту область.
Эпилог
На простых для восприятия примерах мы рассказали о некоторых задачах, которые биоинформатики выполняют очень быстро, если на пороге вирус, захватывающий весь мир. Однако в действительности эти и многие другие задачи мы решаем каждый день. Никакая пандемия не отменяет того, что люди страдают от онкологических и аутоиммунных заболеваний, а дети рождаются с тяжелейшими генетическими дефектами. Создание препаратов, способных помочь таким пациентам, ничуть не менее важно, а возникающие при этом задачи обычно даже сложнее и требуют гораздо больше времени на проведение исследований.
Для создания терапии при таких болезнях мы разрабатываем как биологические, так и низкомолекулярные соединения. Первые требуют генетические конструкции, оптимизацию работы живых клеток и определение сложных физико-химических характеристик получающихся белков и других биологических молекул (например, мы делаем терапевтические вирусы для лечения орфанных заболеваний). Для вторых появляются проблемы течения органического синтеза, проницаемости клеточных мембран, стабильности в различных фракциях микросом и так далее. Чтобы создать такие большие и малые молекулы, мы применяем методы рационального дизайна на основе трехмерных полноатомных моделей молекул и их мишеней, рассчитываем способы связывания терапевтических молекул с их мишенями, оптимизируем схемы химического синтеза, предсказываем зависимость поведения препарата в организме в зависимости от роста и веса пациента и многое другое.
Разумеется, для создания лекарственных средств нужны высококлассные специалисты, как в биологических и химических «мокрых» лабораториях, так и в «сухих» областях вычислительной биологии и биоинформатики. В России нет вузов, которые выпускают готовых вычислительных биологов, ориентированных на решение задач современной разработки лекарственных средств. Долгое время мы обучали специалистов внутри BIOCAD и вырастили не одно поколение востребованных специалистов. Теперь же пришло время открыть полноценную программу по обучению биоинформатике, и преподаваться она будет магистрантам Питерской Вышки. Старт программы — сентябрь 2021 года, но уже в 2020 мы запускаем пилотный трек в рамках уже имеющейся программы «Программирование и анализ данных».
Мы очень ждем студентов с сильным физико-математическим бэкграундом, без химической и биологической подготовки. Преподаватели Питерской Вышки обеспечат лучшие в стране курсы по алгоритмам, программированию, анализу данных, а сотрудники индустрии расскажут спецглавы физики, молекулярной биологии, химии, а также специальные курсы по молекулярным симуляциям, алгоритмам структурной биоинформатики, системной фармакологии и иным важным для области темам. И, что тоже важно, расскажем о настоящих индустриальных биологических задачах и научим использовать полученные знания и навыки для их решения.
Скоро информация о новой программе появится на сайте Санкт-петербургской школы физико-математических и компьютерных наук. Следите за новостями!

shadrap
Спайк белок у второго ковида сложно назвать высоко-консервативным. Научные изыскания говорят о как минимум двух фуриновых сайтах рестрикции, где белок режется, получая затем довольно рандомные инсерции. Собственно говоря они признаны «виновниками» высокой афинности к АСЕ2 и как следствие всем тем «нехорошим» что мы наблюдаем.
Вы как то учитываете эту мутагенную область?
Второй вопрос- почему вакцина, почему не МАБ?
zmactep
Правда, SPIKE не супер-консервативный, но именно он сильнее всего экспонирован на поверхности вируса, а потому является предпочтительной целью для В-клеточного иммунного ответа. Многочисленные секвенирования (придется зарегистрироваться) образцов SARS-CoV-2 из различных пациентов показывают, что текущий мутагенез довольно умеренный и использование RBD оправдано. В случае значительного изменения вируса, конечно, придется делать новую вакцину. Но по накатанным рельсам это может стать рутиной, как ежегодный выпуск противогриппозных вакцин.
Вакцина и моноклональное антитело решают разные задачи. Первая — часть превентивных профилактических мер, второе — уже терапия. Исходный заказ был именно на вакцину. Да и производство антител в требуемых сейчас объемах (если мы говорим о тотальной терапии всех COVID-положительных) задача малореальная. Для тяжелых случаев мы буквально несколько дней назад зарегистрировали anti-IL6R mAb для купирования цитокинового шторма.