
Автор сообщества Фанерозой, биотехнолог, Людмила Хигерович.

На дворе двадцать первый век, стремительными темпами информационные технологии захватывают все больше сфер нашей жизни, включая науку. С каждым годом они все глубже проникают в различные отрасли науки, способствуя их развитию и порождая новые, смежные дисциплины. Таковой, например, является геномика.
Геномика — один из разделов генетики, однако много общего имеет с междисциплинарной биоинформатикой, разделяя с ней предмет и методы исследования. Но прежде, чем мы подробно рассмотрим саму геномику, окунемся в историю ее происхождения.
Точной даты рождения геномика не имеет. Однако ее появление относят к 1980-м годам, незадолго после открытия структуры ДНК и начала распространения методов секвенирования. Некоторые ученые, правда, все же указывают год рождения геномики — 1977, год полной расшифровки генома бактериофага Φ-X179.
Вероятно, тут стоит притормозить и добавить парочку словарных справок под спойлер. Не все наши читатели хорошо знакомы с терминами, а некоторые уже успели позабыть.
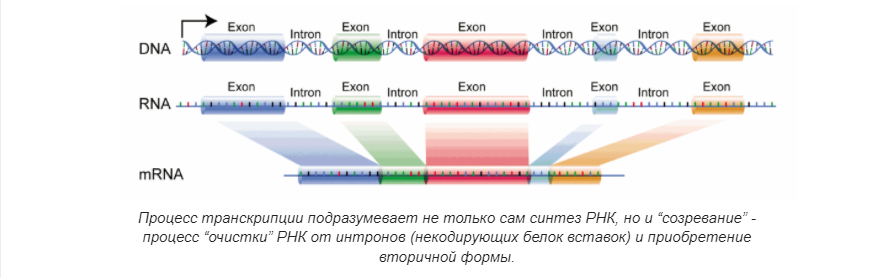
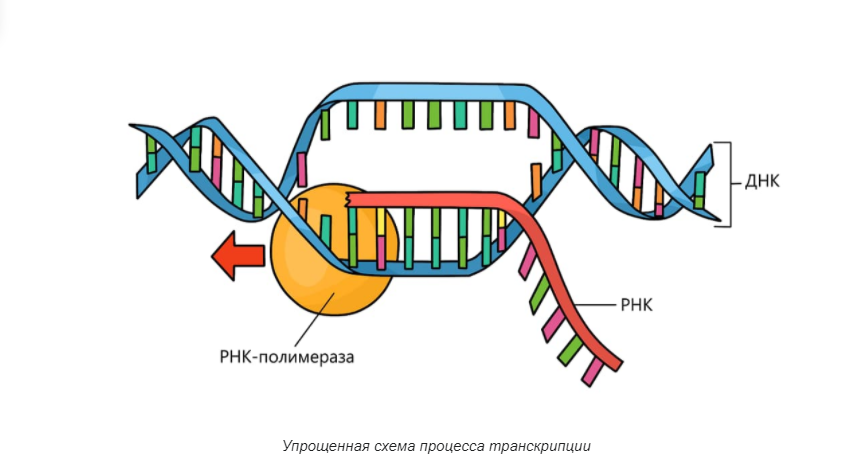
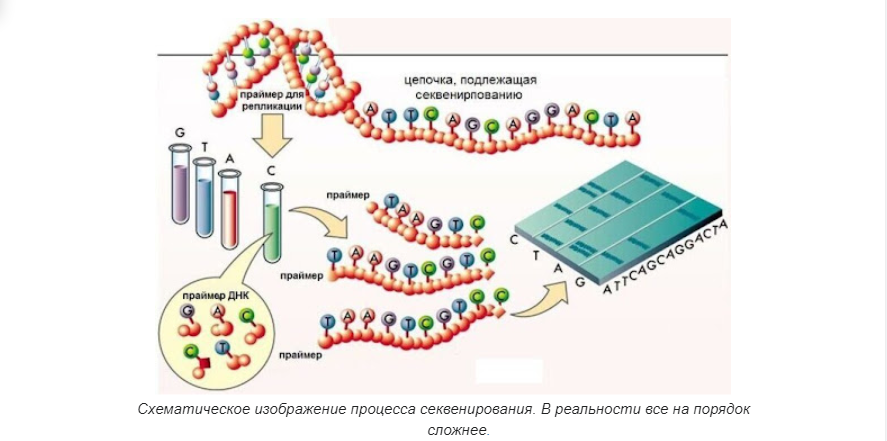
Повторение — мать учения. Итак, как мы говорили выше, геномика — это междисциплинарная область биологии, в которой основное внимание уделяется структуре, функциям, эволюции, картированию и редактированию геномов. Геном — это полный набор ДНК организма, включая все его гены, а также его иерархическую трехмерную структурную конфигурацию. В отличие от генетики, которая относится к изучению отдельных генов и их роли в наследовании, геномика направлена на коллективную характеристику и количественную оценку всех генов организма, их взаимосвязей и влияния на организм.
Почему геномика — важный раздел науки?
Гены могут управлять производством белков, с помощью ферментов и молекул-мессенджеров. В свою очередь, белки составляют структуры тела, такие как органы и ткани, а также контролируют химические реакции и передают сигналы между клетками. Геномика также включает в себя секвенирование и анализ геномов с использованием высокопроизводительного секвенирования ДНК и биоинформатики для сборки и анализа функции и структуры целых геномов. Достижения в области геномики вызвали революцию в исследованиях, основанных на открытиях, и в системной биологии, чтобы облегчить понимание даже самых сложных биологических систем, таких как мозг.
Эта область знаний также включает исследования внутригеномных (внутри генома) явлений, таких как эпистаз (влияние одного гена на другой), плейотропия (один ген влияет на более чем один признак), гетерозис (сила гибрида) и другие взаимодействия между локусами и аллелями внутри генома.

Чем больше люди изучали генетический аппарат живых организмов, тем становилось яснее, что усилий одного только человеческого мозга недостаточно для полноценного анализа и расшифровки. Так что как только появилась возможность машинного анализа, биологи с радостью поручили часть своей работы компьютерам. Так зародилась другая междисциплинарная область знаний — биоинформатика.
Биоинформатика — часть науки, объединяющая в себе генетику, биохимию и молекулярную биологию, математическую статистику и отчасти задевающую краем эволюционную и популяционную биологии, медицину и математическую статистику. Биоинформатики является междисциплинарным полем, которое разрабатывает методы и программные средства для понимания биологических данных, в частности, когда наборы данных являются большими и сложными. Как междисциплинарная область науки, биоинформатика сочетает в себе биологию, информатику, информационную инженерию, математику и статистику для анализа и интерпретации биологических данных. Биоинформатика использовалась для анализа in silico (на компьютере) биологических запросов с использованием математических и статистических методов.
Некоторые соотносят биоинформатику с геномикой, однако они не перекрывают друг друга полностью — скорее, геномика пользуется достижениями биоинформатики, и в то же время сама является инструментом ее пополнения и расширения.
Разделы геномики
За почти пятьдесят лет своего существования геномика значительно расширилась и приобрела внутреннюю структуру — разделилась на несколько направлений, тем не менее, все еще зависящих друг от друга. Деление это весьма условно, многие отделы не просто пересекаются, а значительно перекрывают друг друга.

Геномику подразделяют на следующие направления:
- Структурная геномика
- Функциональная геномика
- Сравнительная геномика
- Музеогеномика
- Когнитивная геномика
- Вычислительная геномика
Структурная геномика — самый крупный и самый “развитый” раздел. Ее задача — выяснение конечной структуры белка, закодированного в исследуемой последовательности. Это важно при расшифровке генетической информации: мало выяснить, какие основания и как расположены в участке ДНК, гораздо более интересно, во что она транскрибируется.
И этот интерес отнюдь не праздный — состав и структура белка могут многое сказать о его функции и месте в метаболизме.

После синтеза аминокислотная цепочка (первичная структура) “плавает” в цитоплазме, и сворачивается в петли. Когда подходящие участки белка сближаются, в дело вступают молекулярные химические силы — некоторые места “слипаются” под действием сил водородных связей, ионных связей, а также сил Ван-дер-Ваальса, образуя вторичную объемную структуру — альфа-спираль и бета-лист. На этом их преобразование не останавливается — в конечном итоге белок приобретает форму глобулы — плотно свернутого клубка. В таком виде он обычно и существует, выполняя свои функции.
Знание третичной структуры позволяет сделать предположение о функциях белка — это не просто комок аминокислот, глобула устроена так, что у нее есть домены — участки с разным предназначением. Условно их делят на основную часть и функциональную — например, ферментирующая ямка (энзимы и пищеварительные ферменты), “хвост” для закрепления на мембране (кинезины, мембранные белки), участки, “примагничивающиеся” к другим белкам (актино-миозиновый комплекс в мышцах) и т. д.
Этому даже посвящен значительный раздел биоинформатики — он так и называется, предсказание структуры белка (protein structure prediction). Это преимущественно математическое моделирование с опорой на молекулярную биологию и химию. Помимо расшифровки первичной структуры, необходимо учитывать две важные вещи — количество свободной энергии и нахождение глобального минимума энергии. Это звучит, как магическая формула, однако на практике это то, от чего зависит, как и насколько преобразуется и свернется белок. Кроме того, требуются огромные вычислительные мощности для расчета всех возможных вариантов пространственной структурой белка — а это миллионы вариантов для белка длиной в сотню аминокислотных остатков. Круг поиска несколько сужают методы предсказания укладки (альфа-спираль или бета-лист, и участки с повышенной вероятностью “слипания”) и гомологическое (сравнительное) моделирование, основанное на знании о структуре хорошо изученных белков. В то же время его осложняют возможные посттранскрипционные преобразования — белок не просто меняет свою пространственную структуру, но также может распадаться, разрезаться ферментами, сшиваться и приобретать конечные свойства только в четвертичной структуре — в комплексе с другим белком.
Помимо этого, структурная геномика в комплексе с функциональной может предсказывать болезни, возникающие при генетических аномалиях. Так, было выяснено, что мутация в одном из белков мембраны клеток может вызывать иммунитет организма к ВИЧ СПИДу, но при этом ослабляет его перед ОРВИ и лихорадкой западного Нила. Этот же белок отчасти ответственен за тип вашего темперамента — гипертимный ли, флегматичный и т. д. Эта часть соотносится с функциональной геномикой.

Функциональная геномика своей основной задачей ставит прослеживание полного пути реализации генома. Она показывает, как определенный ген превращается в белок, и какой конечный признак в организме от него зависит, чем приближается к протеомике — междисциплинарной отрасли науки, изучающей структуру и функции протеинов и белков в частности. Это весьма сложно в рамках многоклеточного организма, так как один и тот же белок выполняет множество функций. Кроме того, разные белки могут одновременно влиять на один и тот же признак.
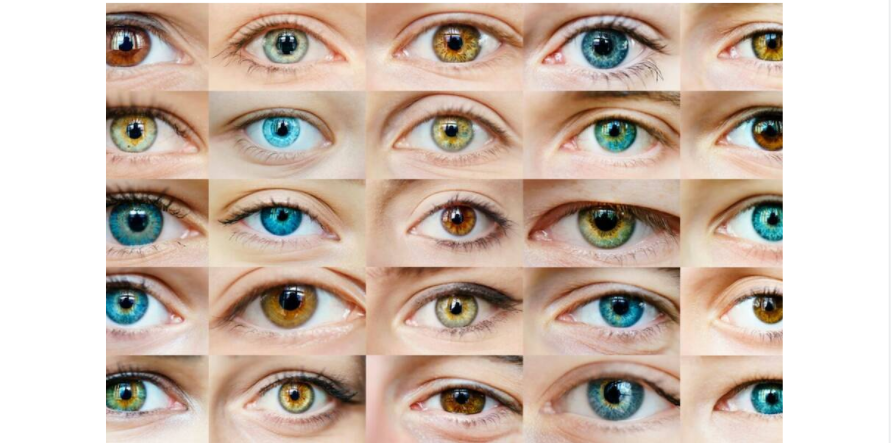
Так, например, цвет глаз человека кодируют как минимум 6-8 генов на 2-3 хромосомах. За светлые глаза в основном отвечает мутация гена OCA2. За синий или зелёный цвет отвечает ген EYCL1 хромосомы 19; за коричневый — EYCL2; за коричневый или синий — EYCL3 хромосомы 15. Кроме того, с цветом глаз связаны гены OCA2, SLC24A4, TYR, влияющие на оттенки и переходные цвета. Все эти гены, вернее, их белки, действуют одновременно, благодаря чему найти два одинаковых рисунка радужки почти невозможно. Кроме того, в зависимости от перенесенных заболеваний, питания и выцветания на солнце цвет глаз меняется в течении всей жизни, что еще больше осложняет работу по составлению статистических исследований.
Сравнительная геномика близка к эволюционной биологии и популяционной генетике. Она сравнивает организацию и функционирование генома у разных систематических групп организмов, изучает их принципиальные сходства и различия, и ищет аналогию между ними. Значительную часть составляет поиск гомологий и аналогий у разных организмов с геномом человека — вопрос больше практический, чем праздный. От этого зависит выбор организма для производства лекарственных средств или даже выращивания органов и тканей для трансплантации. Так, многие фармацевтические препараты, а также витамины производят с помощью бактерий, так как их генетический аппарат почти не преобразует конечный продукт, и при этом обладает достаточным функционалом для синтеза. Ну и, конечно, их относительно легко модифицировать и культивировать.
Музеогеномика — во многом схожа со сравнительной. Только специализируется на исследовании генома музейных экспонатов — палеонтологических, зоологических, ботанических и других. Она позволяет установить степень родства вымерших таксономических групп, проследить эволюцию признака у ископаемых растений и даже предположить, какие животные были предыдущими носителями вирусов и прочих паразитов. Кроме того, музеогеномика позволяет проследить токсигенное влияние человека, сравнивая образцы столетней давности с сегодняшними обитателями Земли.

Когнитивная геномика. Несколько странное направление в контексте современных исследований. Казалось бы, человечество еще в прошлом веке пережило предрассудки о наследовании характера и умственных способностей и связи этого с внешностью. Однако полностью отрицать влияние генетики на мозговую активность — тоже неправильно. Белковый состав мембран нервных клеток, а также степень и качество миелинизации (образования изолирующих оболочек) оказывает сильное влияние на тип нервной системы и темперамент.
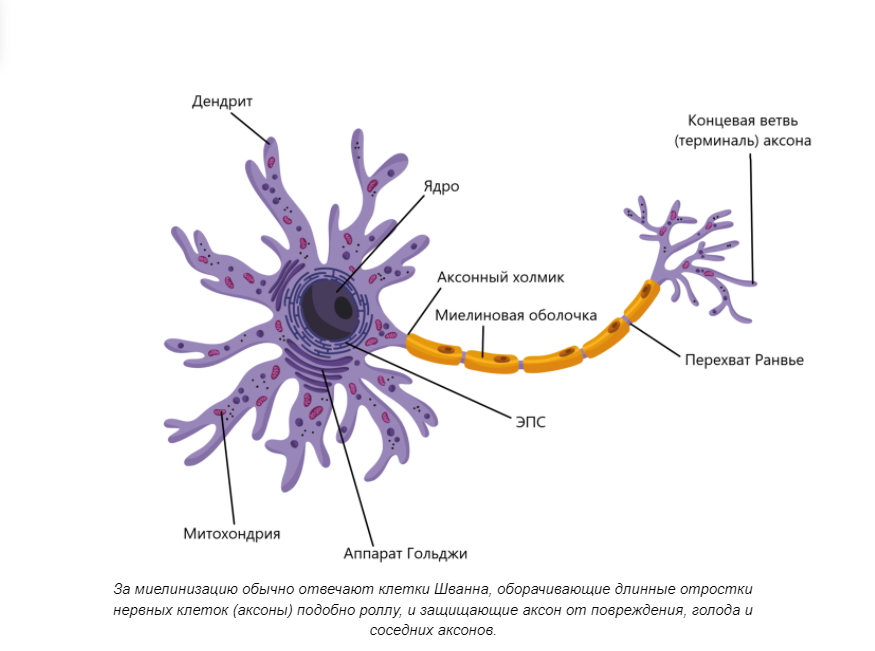
Темперамент человека зависит от типа нервной системы, что в свою очередь зависит от состава и количества мембранных белков, структура которых закодирована в ДНК — по оценке Вайнбергера, примерно 70% всех генов организма оказывает влияние на нервную систему, в частности на головной мозг. С возрастом, разумеется, на темперамент оказывает влияние опыт, физиологические преобразования и воспитание, что формирует вторичные структуры — характер и личность. Однако базовые параметры так или иначе заложены генетически.
В рамках когнитивной отлично уживаются элементы сравнительной геномики. Ученые сравнивают геномы нескольких видов, чтобы выявить генетические и фенотипические различия между ними. Наблюдаемые фенотипические характеристики, связанные с высшей нервной деятельностью, включают поведение, личность, нейроанатомию и невропатологию. Теория когнитивной геномики основана на элементах генетики, эволюционной биологии, молекулярной биологии, когнитивной психологии, поведенческой психологии и нейрофизиологии.
Но когнитивная геномика не ставит своей задачей узнать, кто родился с большим шансом стать гением, а кто с меньшим. В первую очередь когнитивная геномика работает с выявлением причин патологий высшей нервной деятельности и аномалий мозга, а также исследует возможные способы их лечения или уменьшения. Большинство поведенческих или патологических фенотипов происходят не из-за мутации только одного гена, а из-за сложной генетической основы. Однако есть некоторые исключения из этого правила, такие как болезнь Гентингтона, которая вызвана одним конкретным генетическим заболеванием. На возникновение нейроповеденческих расстройств влияет целый ряд факторов, генетических и негенетических. Методы когнитивной геномики были использованы для изучения генетических причин многих психических и нейродегенеративных расстройств, включая синдром Дауна, большое депрессивное расстройство, аутизм и болезнь Альцгеймера.
Вычислительная геномика — самая близкая к биоинформатике часть, родившаяся и развивающаяся одновременно с ней. Так или иначе, ее элементы используют все остальные разделы геномики. Ее основа — вычислительный анализ, позволяющий расшифровать не только отдельные участки или гены, но охватить геном целиком, включая не только последовательность нуклеотидов в ДНК, но и синтезированную на ее основе РНК.
Геномика и Big Data
Геном даже одного простейшего организма состоит из тысяч или даже десятков тысяч пар оснований. Анализ одной только цепочки ДНК из одной хромосомы “вручную” занимает годы, если не десятилетия. Прибавим к этому то, что секвенирование зачастую предполагает разрезание ДНК на маленькие кусочки, и получим еще одну задачку — собрать расшифрованные кусочки в нужном порядке. Эта задача, называемая генетическим картированием, поистине титаническая. И хоть без человеческого умственного труда все равно не обойтись при финальном сведении данных и написании вывода, то значительную часть аналитического труда выполняет компьютер.
Специально для целей геномики даже были разработаны особые программные и иерархические средства анализа. Так, например, были разработаны специфические «конвейеры» анализа, неоднократно “прогоняющие” одну и ту же последовательность, а затем повторяющие эти же процедуры с другой, в конечном итоге сравнивая полученные результаты и фиксируя сходства и различия. Обычно это используется для идентификации генов-кандидатов (изменения в них могут вызвать онкологию) и однонуклеотидных полиморфизмов (точечных мутаций, характеризующихся многократным, идущим подряд повторением основания на одной из цепей, что вызывает разрыв между цепями).
Компьютерный анализ также помогает при аннотировании — маркировании генов. Этот процесс необходимо автоматизировать, потому что большинство геномов слишком велики для аннотирования вручную, не говоря уже о необходимости аннотировать как можно больше геномов, поскольку скорость секвенирования перестала быть проблемой. Аннотации стали возможными благодаря тому факту, что гены имеют узнаваемые начальные и конечные области (промоторы и терминаторы, имеющие зачастую схожий или одинаковый состав у разных групп организмов), хотя точная последовательность, обнаруженная в этих областях, может варьироваться между генами.
Кроме аннотирования и анализа отдельного генома, технологии Big data позволяют массово сравнивать геномы особей в популяциях и даже видов. Так появилась, например, вычислительная эволюционная биология. Эволюционная биология — это изучение наследственности, изменчивости и происхождения видов, а также их изменения с течением времени. Информатика помогла эволюционным биологам, позволив исследователям:
- Отслеживать эволюцию большого числа организмов, измеряя изменения в их ДНК, а не только с помощью физической систематики или физиологических наблюдений
- Сравнивать целые геномы, что позволяет изучать более сложные эволюционные события, такие как дупликация генов, горизонтальный перенос генов, а также предсказывать факторы, важные для видообразования бактерий
- Построение сложных вычислительных моделей популяционной генетики для прогнозирования результатов работы системы с течением времени
- Отслеживать и обмениваться информацией о все большем количестве видов и организмов.
В перспективе это поможет составить более подробное и правдоподобное филогенетическое дерево — т. н. древо жизни, отражающее примерную эволюцию живого.

Такие сложности возникают потому, что в процессе эволюции ДНК не только усложняется, но и упрощается, теряя “ненужные” участки в ходе делеций или отбора. Кроме того, существует такое явление, как горизонтальный перенос генов — обмен участками генома между генетически неродственными группами. Так, например, значительная часть интронов у бактерий, растений и животных, включая человека, — это следы заражения вирусами, которые потеряли часть своего генома, ответственную за синтез оболочки вируса и репликацию.
В рамках вычислительной геномики существует также такое понятие, как Интерактом (Interactome) или сети молекулярного взаимодействия. В двух словах, это совокупность локализаций и взаимодействий конкретной молекулы в рамках одной конкретной клетки. Такая модель также может описывать наборы непрямых взаимодействий между генами. Молекулярные взаимодействия могут происходить между молекулами, принадлежащими к разным биохимическим семействам (белки, нуклеиновые кислоты, липиды, углеводы и т. д.), а также внутри каждого семейства. Когда такие молекулы связаны физическими взаимодействиями, они образуют сети молекулярных взаимодействий, которые обычно классифицируются по природе задействованных соединений. Чаще всего, интерактом относится к сети межбелкового взаимодействия (PPI) (PIN) или ее разновидностям.
Это чрезвычайно сложная схема, реализация которой была бы невозможна без компьютерного моделирования. Она настолько сложна технически и важна практически, что в последнее время выделяется в самостоятельную область биоинформатики.
Биоинформатика для всех
Помимо решения сложных узкопрофильных задач, для которых обычно создаются специальные программные методы, существуют и более широкого спектра сервисы.
Так, есть ряд проектов, призванных помочь любому желающему провести быстрый анализ полученного сиквенса или белка — рано или поздно с такой проблемой сталкивается любой биолог-исследователь.
Первым подобным проектом стал алгоритм Нидлмана — Вунша на основе динамического программирования. Он был создан для того, чтобы сравнить наборы последовательностей аминокислот друг с другом при использовании матриц замен, полученных в более раннем исследовании М. Дейхофф.

Позже появился алгоритм BLAST, который позволяет осуществлять быстрый и оптимизированный поиск в базах данных последовательностей генов. BLAST и его модификации — одни из наиболее широко используемых алгоритмов для этой цели.
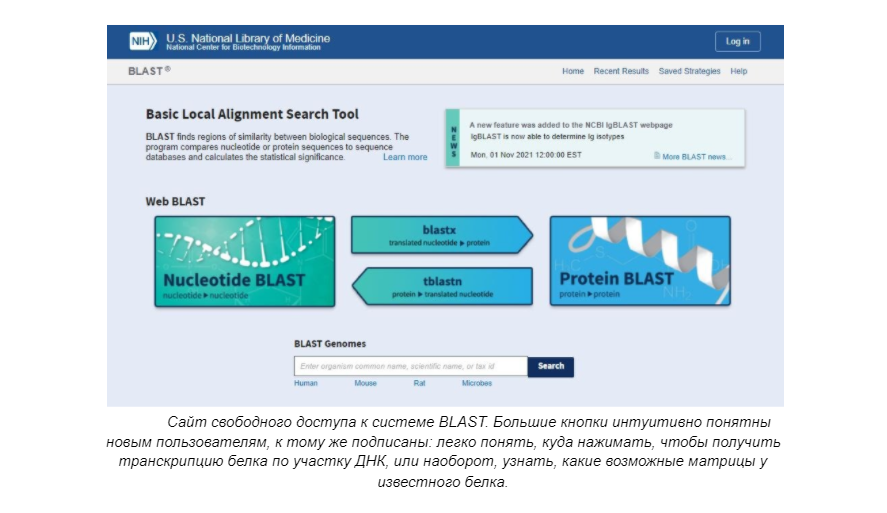
Примерно теми же функциями обладают и системы UGENE и DNAStar, однако пользование ими для обычного человека затруднено, к тому же есть платные пакеты с расширенным функционалом. Кроме того, существуют различные сервисы по моделированию возможной структуры белков. Таковым является, например, Google DeepMind.
На этом мы завершим очередную мини-экскурсию в мир биологической информатики или информативной биологии. Надеемся, что вы сегодня узнали что-то новое и интересное.
Всего хорошего и не болейте!

Lesk AM (2017). Introduction to Genomics (3rd ed.). New York: Oxford University Press. p. 544.
Dong Xu, Lukasz Jaroszewski, Zhanwen Li, Adam Godzik. AIDA: ab initio domain assembly for automated multi-domain protein structure prediction and domain–domain interaction prediction (англ.) // Bioinformatics. — 2015-07-01. — Vol. 31, iss. 13. — P. 2098—2105.
Plomin, R. & Spinath, F.M. (2004). «Intelligence: Genetics, Genes, and Genomics.» Journal of Personality and Social Psychology, 86(1): 112—129
.
Sulem P., Gudbjartsson D. F., Stacey S. N., et al. Genetic determinants of hair, eye and skin pigmentation in Europeans (англ.) // Nat. Genet.: journal. — 2007. — December (vol. 39, no. 12). — P. 1443—1452.
Blue Eyes Versus Brown Eyes: A Primer on Eye Color Архивировано 8 декабря 2012 года… Eyedoctorguide.com. Retrieved on 2011-12-23.
Gonnet G. H. 2012. Surprising results on phylogenetic tree building methods based on molecular sequences. BMC Bioinformatics, 13:148
Google's DeepMind predicts 3D shapes of proteins, The Guardian. 2018
Hug, L., Baker, B., Anantharaman, K. et al. A new view of the tree of life. Nat Microbiol 1, 16048 (2016).
Julian Vosseberg, Jolien J. E. van Hooff, Marina Marcet-Houben, Anne van Vlimmeren, Leny M. van Wijk, Toni Gabaldón & Berend Snel. Timing the origin of eukaryotic cellular complexity with ancient duplications // Nature Ecology and Evolution. 2020. DOI: 10.1038/s41559-020-01320-z.
Papanikolaou, N.; Pavlopoulos, G.A.; Theodosiou, T.; Iliopoulos, I. Protein-protein interaction predictions using text mining methods (англ.) // Methods: journal. — 2015. — Vol. 74. — P. 47—53.
Mikhail Fursov, Olga Golosova, Konstantin Okonechnikov. Unipro UGENE: a unified bioinformatics toolkit (англ.) // Bioinformatics. — 2012-04-15. — Vol. 28, iss. 8. — P. 1166—1167.
Wang L, Eftekhari P, Schachner D, Ignatova ID, Palme V, Schilcher N, Ladurner A, Heiss EH, Stangl H, Dirsch VM, Atanasov AG. Novel interactomics approach identifies ABCA1 as direct target of evodiamine, which increases macrophage cholesterol efflux. Sci Rep. 2018 Jul 23;8(1):11061.
Kotlyar M, Pastrello C, Pivetta F, Lo Sardo A, Cumbaa C, Li H, Naranian T, Niu Y, Ding Z, Vafaee F, Broackes-Carter F, Petschnigg J, Mills GB, Jurisicova A, Stagljar I, Maestro R, Jurisica I (2015). «In silico prediction of physical protein interactions and characterization of interactome orphans». Nature Methods. 12 (1): 79–84.
Robson JF, Barker D (October 2015). «Comparison of the protein-coding gene content of Chlamydia trachomatis and Protochlamydia amoebophila using a Raspberry Pi computer». BMC Research Notes. 8 (561): 561.
Dawson WK, Maciejczyk M, Jankowska EJ, Bujnicki JM (July 2016). «Coarse-grained modeling of RNA 3D structure». Methods. 103: 138–56.
Комментарии (22)

balu736
27.11.2021 16:08+3Понимаю, что начинаю общаться со специалистом. Вы написали: "Темперамент человека зависит от типа нервной системы, что в свою очередь зависит от состава и количества мембранных белков..."
Замечу, вами упущен огромный пласт промежуточных структур мозга, которые развиваясь с утробы матери, вообще никак генетически не определены. Видимо, вы будете очень возражать, поэтому сразу отошлю вас к профессору Савельеву с его книгой "Морфология мозга". Интереснейшее чтиво.
Было время и я верил, что все определено генетикой.

phanerozoi_evidence Автор
27.11.2021 19:08+31) Тушку размечивают хокс-гены.
2) Морфологию мозга Савельева признаюсь не читал, но очень трудно переосилить себя к прочтению после номинации Савельева на премию ВРАЛ. Ученый во многих вещах заблуждался, за что наши коллеги из портала антропогенез вполне заслуженно его критиковали.
phanerozoi_evidence Автор
27.11.2021 17:24+1Выравнивание в blast ncbi, который по сути и есть blast происходит след. образом: загружается файл fasta и далее можно произвести множественное выравнивание. Выравнивание в ugene— загружается файл fasta и производится множественное выравнивание.

zv347
28.11.2021 15:07-1Мне казалось, что в бласте выравнивание всё-таки парное (локальное), а не множественное. MSA можно сделать в NCBI BLAST, но это уже и не бласт.
phanerozoi_evidence Автор
28.11.2021 15:28NCBI BLAST работает на алгоритмах BLAST. Далее из нашей статьи
Позже появился алгоритм BLAST, который позволяет осуществлять быстрый и оптимизированный поиск в базах данных последовательностей генов. BLAST и его модификации — одни из наиболее широко используемых алгоритмов для этой цели
zv347
28.11.2021 15:53Извините за резкость, но вы точно понимаете, о чем пишете? Потому что ваш ответ не имеет никакого отношения к моему вопросу.
Вы до этого написали, что в бласте делается множественное выравнивание. Я поправил, что не множественное, а парное.phanerozoi_evidence Автор
28.11.2021 15:59В BLAST ncbi делается множественное выравнивание. BLAST ncbi работает на BLAST - basic local alignment serch tool.
zv347
28.11.2021 15:04+1Бородатый биологический анекдот про блохПриходит студент на экзамен, выучил только про блох. Достается билет про рыб. Он начинает отвечать:
— Рыбы покрыты чешуёй. А если бы у них была шерсть — там были бы блохи…
К чему это я. Статья очень поверхностная и неконсистентная, но это, может, и не проблема, смотря какую задачу ставили перед собой авторы. Но такое ощущение, что понятие «геномика» натянули на всё, на что только можно и нельзя. Протеомика и особенно ПТМ, интерактомика, предсказание структуры белков, тем более физиология (а еще есть сигналомика и другие омики) — это отдельные области со своей методологией. Да, не всё можно объяснить с помощью секвенирования и поиска ассоциаций (вот удивительно). Но теперь всю биологию что ли будем называть геномикой?phanerozoi_evidence Автор
28.11.2021 16:03Во-первых, внизу ссылки на источники, во-вторых, структурная геномика занимается предсказанием структуры белков.

Doc_x800
28.11.2021 15:41+1BLAST - basic local alignment serch tool. Как было здесь сказано это алгоритм. Кстати, с кучей параметров. И используется во многих ПРОГРАММНЫХ пакетах для генетики. В том числе и UGENE и DNASTAR и VECTOR NTI SUIT и т.д.
mahajrod
Пожалуйста, уберите это убожество в черновики.
Говорю как биоинформатик.
Написано много чуши.
Особенно про разделы геномики.
Дикие, безумные скачки в повествовании.
Например, пишете про blast как алгоритм, потом внезапно даете скрин страницы ncbi blast, который есть лишь веб обертка над бластом. А затем еще и заявляете, что ugene обладает примерно тем же функционалом.
И такого бреда очень много.
phanerozoi_evidence Автор
Текст был написан при консультации с биоинформатиками. Касаемо обертки, дык с генами работают в визуализации на странице ncbi. В Ugene есть куча всего того, что и есть в blast.
mahajrod
Не похоже. Или получилось из разряда "ученый изнасиловал журналиста". Или уровень компетенции этих биоинформатиков очень низок.
С генами работают многими способами. И не только используя выравнивания. И не только на ncbi. И не только с деятком-другим генов. Мне вот очень любопытно как вы будете работать с десятком тысяч генов "в визуализации на странице ncbi".
Blast - это лишь один сравнительно небольшой, но популярный пакет тулов. Ugene - это по сути обертка и графический интерфейс к куче тулов. Не только к blast. Ugene корректно сравнивать, например, с Genious. Но никак не с бластом.
От этой статьи откровенно бомбит. Мои студенты отчеты лучше пишут.
phanerozoi_evidence Автор
С десятками не работал, но работал с тысячей. Сложно, долго, мучительно, но возможно. Работал с десятком тысяч в mega 10 при нажатии ncbi blast. Там есть эта функция. Комп при этом работает сутками.
К туче таких же как и blast
И? Работают многими способами, где это отрицалось? Вопрос риторический— нигде не отрицалось, это был пример.
С бластом никто не запрещал сравнивать. Мы могли сравнить и с Genious, но решили сравнить с Blast, как и ученые в списках использованной литературы тоже сравнивали. И ничего страшного не произошло.
Аппеляция к личности.
P.S. Не бомбите в комментариях. Напишите опровержение этой статьи на хабре.
mahajrod
Вы сравниваете не сравниваемое и не понимаете этого. Это как сравнивать IDE и библиотеку из какого-нибудь языка.
Вы осознаете отличие между бластом и веб интерфейсом к нему на ncbi?
И аппеляция прежде всего к Вашей квалификации и качеству статьи. Нужно разбираться в области прежде чем писать обзорные статьи, пусть и научпоп.
phanerozoi_evidence Автор
Веб интерфейс бласта представлен для удобства работы с алгоритмом бласт, по сути на ncbi работают на алгоритме blast, при помощи интерфейса. Об этом написано и в списке литературы.
P.S. Мы разбираемся в области в которой пишем. Мы считаем, что ответили максимально развернуто, на абстрактные претензии и мы так и не увидели от Вас компетентных комментариев без переходов на личности. Поэтому мы справедливо считаем, что этот вопрос закрыт. Если же вы хотите поспорить, то напишите опровержение данной статьи. Далее мы не будем кормить вас нашим временем. Всего доброго.
rtakyiv
Справедливости ради, сравнение BLAST с Ugene действительно нелепое. BLAST - это в первую очередь алгоритм (семейство алгоритмов). Ugene - одна из 100500 софтин которые умеют в выравнивания. BLAST появился в 90м и использовался везде и всеми, так что даже в русскоязычном сообществе люди говорят "бластить". Ugene появился в 2008, и его почти не знают.
Не вполне ясно зачем писать коротенькую статейку о филде про который написаны тома и тома. И слог в статье странооватый. Я вот 5 раз читал фразу "Достижения в области геномики вызвали революцию в исследованиях, основанных на открытиях, и в системной биологии, чтобы облегчить понимание даже самых сложных биологических систем, таких как мозг." Это о чем вообще?
phanerozoi_evidence Автор
Ваши части цитат
Не противоречат тому что написано в статье. Все почти тоже самое за некоторым исключением про известность и то, с этим статья не спорит. Спасибо, что процитировали нашу статью своими словами.
Мы действительно знаем что Ugene - одна из 100500 софтин которые умеют в выравнивания, но их умеет делать и интерфейс ncbi, который работает на алгоритмах Blast, о чем написано и на самом сайте. Да и сам Blast делает тоже самое.Поэтому Ваша часть цитаты
скорее ваша цитата выглядит забавно, так как UGENE и DNAStar цитирую:
Касаемо этого:
Статья посвящена далеко не только филду, так как статья посвящена геномике, биоинформатике и немного bigdata. Если об этом читать не прыгая через строчку, то должно быть понятно.
О достижениях в области геномики которые произвели колосальный прорыв в различных исследованиях, которые опирались на открытия в том числе таких дисциплин как кибернетка, системный анализ, системная инженерия, системная нейрофизиология (все что попадает в системную биологию) и тем самым эти достижения облегчают понимание даже самых сложных систем таких как мозг. Ничего сложного для понимания в цитате нет.
rtakyiv
Сложный для понимания там формат изложения материала. Половина предложений грамматически не согласована, вторая половина - не понятно для чего написана. Так пишет gpt3 и люди, когда пишут "на отвали". Это предложение я привел просто как пример. Вы когда пишете, перечитайте что написано. Хорошее правило - если смысл абзаца (предложения) ясен без предложения (слова) - выбрасывайте его. В вашем тексте: достижения вызвали революцию в науках основанных на открытиях. Чтобы облегчить понимание. Мозг. Это же вода.
Не имею ничего против вас лично или вашегл коллектива как автора(ов), не исключаю что у вас есть другие интересные и хорошо написанные статьи. Но этот конкретный материал - без смысла.
phanerozoi_evidence Автор
Любое мнение, важно. Спасибо за критику.
shadrap
Не понимаю, в чем тут спор и недовольство - Бласт родился как алгоритм, сейчас это вполне юзабельный и насыщенный тулс, думаю что более часто используемый чем Юген. Юген , родился как супер-мульти емкий тулс, включающий линк к тому же бласту, гораздо дальше выходящий за рамки выравнивания. Конструирование генов, чуть менее удобно, субъектино, чем в Геном Эдитор, но зато с воркфлоу, опять же тулс для работы с HMMER - да много чего, не представляю даже зачем столько собрано в одном месте.
Для меня Бласт прежде всего некое родство тулса и базы нцби.
phanerozoi_evidence Автор
Да я сам если честно не понимаю недовольства. Об этом же написано в статье, а ugene вообще как пример программы приведен. Как видно из этого комментария
говоря на сленге, критики не выкупили статью из-за стиля написания.