
Сегодня публикуем последнюю задачу из цикла, в котором мы рассказываем, как работать с генетическими данными. Первая и вторая задачи уже опубликованы: их можно решить и прислать ответы нам. Предупреждаем, что эта задача занимает больше времени, чем остальные.
Главный приз — Полный геном.

Ранее мы уже делились полезной информацией и ссылками, которые могут пригодиться для работы с биоинформатическими данными. Советуем сначала прочитать предыдущие статьи, если вы их пропустили:
Что такое Полный геном и зачем он нужен
Задача №1. Узнайте пол и степень родства.
Задача №2. Определение популяционной структуры
Дисклеймер
Работа с генетическими данными проводится на Unix системах (Linux, macOS), так как некоторые команды и ПО недоступны на Windows. Поэтому для пользователей Windows одним из самых простых решений будет арендовать виртуальную машину с Linux.
Все описанные ниже операции выполняются в командной строке — терминале. Перед началом выполнения узнайте, как работать в терминале под управлением вашей ОС и использовать команды, так как некоторые из них могут потенциально навредить ОС и вашим данным.
Мы собрали образ виртуальной машины (ВМ) со всем необходимым ПО на Яндекс.Облаке. Инструкции по настройке ВМ и установке ПО можно найти в статье с первой задачей. Там же есть инструкция, как настроить машину, чтобы пользоваться ею бесплатно до 31 декабря 2019 года.
В этой задаче нужно сконвертировать данные генотипирования из формата VCF в формат 23andMe, загрузить полученные файлы в сервис Promethease и ознакомиться с содержимым отчета для каждого образца.
Для выполнения задачи понадобится программный пакет BCFtools. Если вы его еще не установили, прочитайте статью с первой задачей. В ней содержатся инструкции по установке. Напоминаем, что для участия в новогоднем конкурсе 2019 необходимо выполнение всех задач.
Кроме BCFtools, вам понадобится файл
Существует много сервисов, которые проводят анализ данных генотипирования: MyHeritage, Promethease, FamilyTreeDNA, DNA.LAND, GEDmatch. Они дают загрузить данные генотипирования в различных форматах, зачастую специфичных для конкретного поставщика услуг генотипирования (Ancestry, 23andMe, MyHeritage, FamilyTreeDNA, GenesForGood и другие). Наиболее лояльным к формату данных является Promethease: в данный сервис можно загрузить как VCF, так и файлы в формате 23andMe.
Существует несколько проблем совместимости форматов и сервисов:
Напоминаем, что в данном руководстве используются специально отобранные нами открытые данные из проекта «1000 Геномов». Для анализа мы выбрали 10 образцов с информацией о генотипах ~85 миллионов вариаций, которые получены путем анализа данных NGS с выравниванием на версию генома GRCh37. Родственные связи и популяции данных образцов указаны на Рисунке 1.

Рисунок 1. Родословная используемых в VCF образцов (квадрат соответствует мужскому полу, круг — женскому). Пунктирная линия соответствует неустановленному родству второго порядка.
Ниже приведены инструкции по конвертации VCF файла и загрузке полученных данных в сервис Promethease, который совсем недавно стал бесплатным. Предлагаем ознакомиться с полученным отчетом Promethease по какому-либо из образцов. Используйте отфильтрованный по списку вариаций VCF файл, полученный в задаче №1.
Команда
Скрипт
Перейдите на promethease.com и зарегистрируйтесь. Нажмите на кнопку Upload raw data (Рисунок 2) и загрузите файл
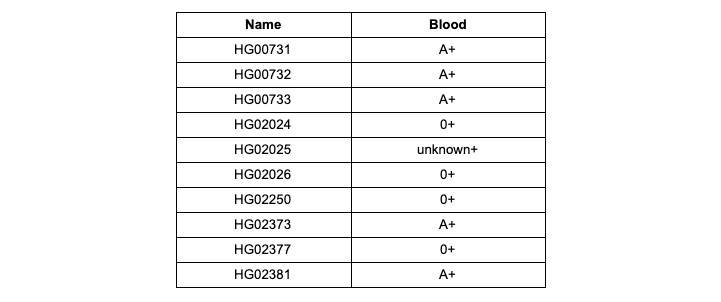
Таблица 1. Группы крови и Резус-фактор, полученные по результатам анализа Promethease образцов из демонстрационного датасета
Отметим, что используемые в данном руководстве файлы всегда содержат заполненное поле ALT (альтернативный аллель), что позволяет понять, к какому типу относится каждая из вариаций (INS, DEL, SNV) и правильно создать запись в формате 23andMe. Данные полногеномного секвенирования в Атлас содержат заполненный ALT аллель только в тех местах, где этот аллель был обнаружен, в противном случае информации для заполнения поля ALT при генерации VCF файла просто не существует. Выдача данных по гомозиготным референсным сайтам (позициям в геноме, где не был обнаружен референсный аллель) необходима, так как клинический эффект имеют не только обнаруженные вариации нуклеотидной последовательности, но и их отсутствие.
Отсутствие ALT аллеля в таких позициях генома не позволяет определить тип генетической вариации, для которой был обнаружен только референсный (REF) аллель. Запись генотипов для таких случаев усложняется необходимостью использовать источник информации о возможных аллелях для данной вариации и не покрывается данным руководством. Если потенциально вы будете использовать данное руководство и скрипт
Для того чтобы скрипт
на
Эта замена позволит выводить в
Для того, чтобы получить отчет Promethease по полному VCF файлу полногеномного секвенирования в Атлас, можно загрузить в Promethease собственно сам VCF файл, однако нужно иметь ввиду, что размер предоставляемого Атлас сжатого VCF файла — около 8 гигабайт, тогда как Promethease позволяет загружать файлы не более 4 гигабайт. Описание решений этой проблемы доступно по ссылке. Еще одно решение заключается в разделении VCF файла на несколько частей (меньше 4 гигабайт каждая) и загрузке каждой в качестве дополнительного файла в меню загрузки данных Promethease.
Загрузите сконвертированные данные каждого из 12 образцов тестового датасета, который вы отфильтровали по списку вариаций в первой задаче, в Promethease и составьте таблицу соответствия идентификатор образца — определенная системой интерпретации Promethease группа крови AB0/Rh (Rh — Резус-фактор). Группы крови, определенные вероятностно и записанные с приставкой «prob» в отчете Promethease, запишите без приставки. Неопределенные значения запишите как unknown (Резус-фактор для unknown групп крови по-прежнему необходимо записать, если он определен). Пример представлен в Таблице 1.
Конвертация VCF в используемый выше формат в предложенной реализации сильно упрощена, но требует значительного количества времени. Для оптимизации вы можете написать скрипт с циклом, который автоматически будет выполнять генерацию этих данных, итерируя по набору идентификаторов. Можно сделать несколько таких скриптов и каждому передать разные наборы идентификаторов образцов для параллельного выполнения, однако количество параллельно запущенных скриптов не должно превышать количество ЦПУ вашего компьютера/виртуальной машины. Хорошее описание создания таких циклов доступно по ссылке. При работе на Яндекс.Облаке вы можете при необходимости создать еще одну виртуальную машину с бо́льшим количеством виртуальных ЦПУ, что позволит пропорционально сократить время на выполнение задачи.
Это последняя задача из нашего цикла. Ответы присылайте на почту wgs@atlas.ru до 26 декабря до 23:59. Правильные ответы и имена победителей опубликуем 28 декабря. Победитель получит тест Полный геном, а второе и третье места — генетический тест «Атлас». Также будут специальные призы от Яндекс.Облако. Бывшие и настоящие сотрудники Атласа в конкурсе не участвуют ;)
Главный приз — Полный геном.
Ранее мы уже делились полезной информацией и ссылками, которые могут пригодиться для работы с биоинформатическими данными. Советуем сначала прочитать предыдущие статьи, если вы их пропустили:
Что такое Полный геном и зачем он нужен
Задача №1. Узнайте пол и степень родства.
Задача №2. Определение популяционной структуры
Дисклеймер
Работа с генетическими данными проводится на Unix системах (Linux, macOS), так как некоторые команды и ПО недоступны на Windows. Поэтому для пользователей Windows одним из самых простых решений будет арендовать виртуальную машину с Linux.
Все описанные ниже операции выполняются в командной строке — терминале. Перед началом выполнения узнайте, как работать в терминале под управлением вашей ОС и использовать команды, так как некоторые из них могут потенциально навредить ОС и вашим данным.
Необходимое ПО
Мы собрали образ виртуальной машины (ВМ) со всем необходимым ПО на Яндекс.Облаке. Инструкции по настройке ВМ и установке ПО можно найти в статье с первой задачей. Там же есть инструкция, как настроить машину, чтобы пользоваться ею бесплатно до 31 декабря 2019 года.
В этой задаче нужно сконвертировать данные генотипирования из формата VCF в формат 23andMe, загрузить полученные файлы в сервис Promethease и ознакомиться с содержимым отчета для каждого образца.
Формат 23andMe представляет собой текстовый формат хранения данных генотипирования и содержит 4 поля, разделенных табуляцией. Первое поле содержит идентификатор вариации (например, rsID), второе — хромосому (допустимые значения этого поля 1-22, Х, Y и MT), третье — позицию на хромосоме, четвертое — генотип (диплоидный при наличии двух гомологичных хромосом, гаплоидный в других случаях). Этот формат поддерживается многими сервисами интерпретации, поэтому в задаче мы будем работать именно с ним.
Для выполнения задачи понадобится программный пакет BCFtools. Если вы его еще не установили, прочитайте статью с первой задачей. В ней содержатся инструкции по установке. Напоминаем, что для участия в новогоднем конкурсе 2019 необходимо выполнение всех задач.
Кроме BCFtools, вам понадобится файл
create_23andme.sh — bash-скрипт, который используется для генерации данных в формате 23andMe. Этот файл расположен в каталоге /Technical как на Яндекс.Облаке, так и в архиве для скачивания, доступном по ссылке в статье.Взять на заметку
Существует много сервисов, которые проводят анализ данных генотипирования: MyHeritage, Promethease, FamilyTreeDNA, DNA.LAND, GEDmatch. Они дают загрузить данные генотипирования в различных форматах, зачастую специфичных для конкретного поставщика услуг генотипирования (Ancestry, 23andMe, MyHeritage, FamilyTreeDNA, GenesForGood и другие). Наиболее лояльным к формату данных является Promethease: в данный сервис можно загрузить как VCF, так и файлы в формате 23andMe.
Существует несколько проблем совместимости форматов и сервисов:
- Разные компании используют разные версии генома для картирования генетических вариаций.Эта проблема решается так называемым лифтовером (liftover), когда происходит замена позиций генетических вариаций в исходных данных на соответствующие им в другой версии генома. Например, Атлас выдает данные генотипирования по версии генома GRCh38, а GEDmatch принимает данные по предыдущей версии генома GRCh37. Конвертация координат генетических вариаций из GRCh38 в GRCh37 и называется лифтовером.
- Использование уникальных идентификаторов генетических вариаций, отличных от rsIDs. Подобные несовместимости решаются путем исключения подобных записей из файла либо их аннотированием — присвоением идентификатора rsID. Второе возможно не всегда.
- Сервисы используют фиксированный набор генетических вариаций. Иногда несоответствие хотя бы части загружаемых данных приводит к ошибке загрузки. Данная проблема актуальна, например, для MyHeritage. Ее можно решить путем выделения набора идентификаторов генетических вариаций, не вызывающих ошибку загрузки.
Используемые данные
Напоминаем, что в данном руководстве используются специально отобранные нами открытые данные из проекта «1000 Геномов». Для анализа мы выбрали 10 образцов с информацией о генотипах ~85 миллионов вариаций, которые получены путем анализа данных NGS с выравниванием на версию генома GRCh37. Родственные связи и популяции данных образцов указаны на Рисунке 1.
Рисунок 1. Родословная используемых в VCF образцов (квадрат соответствует мужскому полу, круг — женскому). Пунктирная линия соответствует неустановленному родству второго порядка.
Конвертация VCF
Ниже приведены инструкции по конвертации VCF файла и загрузке полученных данных в сервис Promethease, который совсем недавно стал бесплатным. Предлагаем ознакомиться с полученным отчетом Promethease по какому-либо из образцов. Используйте отфильтрованный по списку вариаций VCF файл, полученный в задаче №1.
# перейдите в нужную вам директорию
bcftools query -s HG00731 -f '[%SAMPLE]\t%ID\t%CHROM\t%POS\t%REF\t%ALT\t[%GT]\n' -e '%ID=="."' CEI.1kg.2019.demo.subset.vcf.gz | create_23andme.sh > HG00731.subset.23andme.txt
# скачайте на локальный компьютер файл HG00731.subset.23andme.txtКоманда
bcftools query позволяет извлекать из VCF файла любую доступную информацию в формате, заданном пользователем после флага -f. Флаг -s указывает на идентификатор образца (HG00731), для которого необходимо извлечь данные. Флаг -е используется для обозначения критерия исключения, в данном случае ’%ID=="."’ исключает записи, в которых нет идентификатора rsID. Выход с bcftools query передается на вход в скрипт create_23andme.sh, который преобразует данные в TSV формат с 4 колонками (rsID, хромосома, позиция, генотип) и записывает их в файл. Можете скачать и сохранить скрипт create_23andme.sh себе для работы с собственными данными полногеномного секвенирования.Скрипт
create_23andme.sh использует извлеченные из VCF файла поля для определения типа генетической вариации (однонуклеотидная вариация SNV, инсерция INS или делеция DEL) и записи идентификатора rsID, хромосомы, позиции и аллелей в stdout в соответствии с определенным типом вариации (A, G, T и С являются допустимыми аллелями для типа SNV, I и D — допустимые обозначения аллелей для типов INS и DEL).Имейте в виду, что процесс конвертации занимает достаточно много времени: около 4 часов на один файл для одного образца с ~1 миллионом вариаций. Параллелизация BCFtools не поддерживается.
Перейдите на promethease.com и зарегистрируйтесь. Нажмите на кнопку Upload raw data (Рисунок 2) и загрузите файл
HG00731.subset.23andme.txt. После окончания загрузки нажмите на кнопку Create free report и введите желаемое название отчета, который будет сформирован по вашим данным. После составления отчета вам придет уведомление на электронную почту и вы сможете ознакомиться с содержимым отчета. Найдите в отчетах для каждого образца группу крови, которую определила система интерпретации Promethease, в системе AB0/Rh (Rh — Резус-фактор). Проверьте полученные вами результаты на соответствие с Таблицей 1.Таблица 1. Группы крови и Резус-фактор, полученные по результатам анализа Promethease образцов из демонстрационного датасета
Атлас использует отличающиеся от Promethease пороги включения того или иного признака в интерпретацию по уровню доказательности. Под уровнем доказательности понимается совокупность результатов статистических тестов и критериев значимости каждой связи, наблюдаемой между генетической вариацией и какой-либо чертой человеческого организма. Множество признаков, которые можно найти в отчете Promethease, имеют низкий уровень доказательности и/или имеют высокий уровень только в ограниченном наборе популяций, например только для представителей азиатской популяции.
На заметку
Опытным путем нами был установлен основанный на чипе Infinium Global Screening Array v2.0 список генетических вариаций, которые могут быть загружены в MyHeritage. Этот список (
external_interpretation_rsids.txt) хранится в отдельном файле в каталоге /Technical, и его можно использовать для фильтрации VCF с последующей конвертацией по аналогии с инструкцией выше. Также этот файл можно использовать для фильтрации данных генотипирования с чипа, чтобы иметь возможность загрузить их на MyHeritage. Если у вас уже есть приобретенный генетический тест «Атлас», вы можете выгрузить данные генотипирования в формате из личного кабинета и отфильтровать их по предложенному списку вариаций — первая колонка в выгружаемых из личного кабинета данных.Отметим, что используемые в данном руководстве файлы всегда содержат заполненное поле ALT (альтернативный аллель), что позволяет понять, к какому типу относится каждая из вариаций (INS, DEL, SNV) и правильно создать запись в формате 23andMe. Данные полногеномного секвенирования в Атлас содержат заполненный ALT аллель только в тех местах, где этот аллель был обнаружен, в противном случае информации для заполнения поля ALT при генерации VCF файла просто не существует. Выдача данных по гомозиготным референсным сайтам (позициям в геноме, где не был обнаружен референсный аллель) необходима, так как клинический эффект имеют не только обнаруженные вариации нуклеотидной последовательности, но и их отсутствие.
Отсутствие ALT аллеля в таких позициях генома не позволяет определить тип генетической вариации, для которой был обнаружен только референсный (REF) аллель. Запись генотипов для таких случаев усложняется необходимостью использовать источник информации о возможных аллелях для данной вариации и не покрывается данным руководством. Если потенциально вы будете использовать данное руководство и скрипт
create_23andme.sh для конвертации VCF файла, полученного после полногеномного секвенирования в Атлас, сконвертированный файл не будет содержать референсные гомозиготные генотипы, так как скрипт create_23andme.sh явным образом фильтрует такие записи для исключения ошибок при формировании записей для инсерций и делеций.Для того чтобы скрипт
create_23andme.sh все же выдавал референсные гомозиготные генотипы, вам необходимо заменить в нем содержимое строк 25–28...
if [ "$ALT" == "." ] || [[ "$ALT" == *"*"* ]]
then
continue
fi
...на
...
if [[ "$ALT" == *"*"* ]]
then
continue
fi
if [ "$ALT" == "." ]
then
echo -e "$RSID\t$CHR\t$POS\t$REF$REF"
fi
...Эта замена позволит выводить в
stdout записи с гомозиготными референсными генотипами. Необходимо иметь в виду, что такие записи для инсерций и делеций будут некорректны, так как допустимые аллели в используемом формате для инсерций и делеций — это I и D, а скрипт будет использовать аллели A, G, T или C. Чтобы правильно выводить данные для инсерций и делеций, необходимо заранее знать о том, какой тип вариации свойственен данной позиции генома, в которой не был обнаружен ALT аллель. Эту информацию можно получить путем анализа ALT аллеля при его наличии (уже реализовано в create_23andme.sh) или использованием внешней базы данных, например, dbSNP (нет в create_23andme.sh).Для того, чтобы получить отчет Promethease по полному VCF файлу полногеномного секвенирования в Атлас, можно загрузить в Promethease собственно сам VCF файл, однако нужно иметь ввиду, что размер предоставляемого Атлас сжатого VCF файла — около 8 гигабайт, тогда как Promethease позволяет загружать файлы не более 4 гигабайт. Описание решений этой проблемы доступно по ссылке. Еще одно решение заключается в разделении VCF файла на несколько частей (меньше 4 гигабайт каждая) и загрузке каждой в качестве дополнительного файла в меню загрузки данных Promethease.
Третье задание конкурса
Загрузите сконвертированные данные каждого из 12 образцов тестового датасета, который вы отфильтровали по списку вариаций в первой задаче, в Promethease и составьте таблицу соответствия идентификатор образца — определенная системой интерпретации Promethease группа крови AB0/Rh (Rh — Резус-фактор). Группы крови, определенные вероятностно и записанные с приставкой «prob» в отчете Promethease, запишите без приставки. Неопределенные значения запишите как unknown (Резус-фактор для unknown групп крови по-прежнему необходимо записать, если он определен). Пример представлен в Таблице 1.
Конвертация VCF в используемый выше формат в предложенной реализации сильно упрощена, но требует значительного количества времени. Для оптимизации вы можете написать скрипт с циклом, который автоматически будет выполнять генерацию этих данных, итерируя по набору идентификаторов. Можно сделать несколько таких скриптов и каждому передать разные наборы идентификаторов образцов для параллельного выполнения, однако количество параллельно запущенных скриптов не должно превышать количество ЦПУ вашего компьютера/виртуальной машины. Хорошее описание создания таких циклов доступно по ссылке. При работе на Яндекс.Облаке вы можете при необходимости создать еще одну виртуальную машину с бо́льшим количеством виртуальных ЦПУ, что позволит пропорционально сократить время на выполнение задачи.
Это последняя задача из нашего цикла. Ответы присылайте на почту wgs@atlas.ru до 26 декабря до 23:59. Правильные ответы и имена победителей опубликуем 28 декабря. Победитель получит тест Полный геном, а второе и третье места — генетический тест «Атлас». Также будут специальные призы от Яндекс.Облако. Бывшие и настоящие сотрудники Атласа в конкурсе не участвуют ;)