
Видимо, астрологи Хабра объявили день Python в биологии. Сегодня мы приготовили для вас материал, в котором аспирант Эрин Уилсон делится кодом умозрительного примера прогнозирования количественной оценки последовательностей ДНК. За подробностями приглашаем под кат — к старту нашего флагманского курса по Data Science.
ДНК — поток комплексных данных, который можно представить в виде нуклеотидной последовательности ACTG. Тем не менее многие сложные паттерны и структурные нюансы трудно понять человеку, который смотрит на необработанную последовательность нуклеотидов.
В последние годы в моделировании ДНК достигли заметного прогресса. Этот прогресс стал возможным благодаря технологиям глубокого обучения.
Для моделирования ДНК исследователи применяли различные методы. В их числе свёрточные нейросети (CNN), сети с долговременной и кратковременной памятью (LSTM) и даже трансформеры для прогноза различных геномных измерений непосредственно по последовательностям ДНК. Самое полезное в этих моделях — автоматический поиск паттернов и мотивов последовательностей ДНК при достаточном количестве качественных обучающих данных. При этом исследователь не должен заранее указывать искомые паттерны. Нейросеть может их прогнозировать. Полезность глубокого обучения в геномике прибавляет энтузиазма всем, кому нужно сопоставлять последовательности ДНК с биологическими функциями!
Как аспирант, увлечённая математическими методами решения задач рационального природопользования и синтетической биологии, я с интересом изучила возможности PyTorch для исследования паттернов последовательностей ДНК.
Обучающих материалов по PyTorch немало, но многие из них посвящены работе с графикой и языковыми данными. ДНК использовалась в качестве входных данных в целом ряде интересных проектов, которые разработали в PyTorch фреймворки для моделирования различных биологических явлений 1, 2, 3, но они могут быть довольно трудными для новичка.
Мне было нелегко найти примеры в PyTorch для новичков, которые также были бы связаны с ДНК, поэтому я составила это краткое руководство на случай, если будущие архитекторы моделей ДНК найдут его полезным для начала работы.
Само руководство можно запустить в интерактивном режиме блокнота Jupyter. Проследить его план можно по краткому описанию ключевых понятий и Github gists в остальной части этой статьи.
Научим решать интересные задачи, чтобы вы прокачали карьеру или стали востребованным IT-специалистом:
- Полный курс по Data Science. Получите одну из самых перспективных профессий за 24 месяца.
- «Fullstack-разработчик на Python». Станьте незаменимым специалистом-универсалом за 15 месяцев.
- «Уверенный старт в IT». Пройдите лучший курс для новичков: попробуйте 9 профессий и освойте подходящую именно вам.

Построим модель для прогноза количественных показателей по последовательности ДНК в PyTorch
В этом руководстве рассказывается, как PyTorch использует необработанные последовательности ДНК в качестве входных данных и вводит их в модель нейросети, чтобы прогнозировать количественный балл по самой последовательности.
План руководства
- Синтез данных ДНК.
- Подготовка данных для обучения в PyTorch.
- Определение модели в PyTorch.
- Определение функций обучающего цикла.
- Запуск моделей.
- Проверка прогнозов модели на тестовом наборе данных.
- Визуализация свёрточных фильтров.
- Итоги.
Полагаю, что читатель знаком со следующими аспектами машинного обучения:
- Нейросеть и свёрточная нейросеть (CNN).
- Эпохи обучения моделей.
- Разделение данных на наборы train/val/test.
- Функции потерь и сравнение кривых потерь train и val.
Также полагаю, что читатель знаком с некоторыми понятиями биологии. Это:
- Нуклеотиды в ДНК.
- Регуляторный мотив.
- Визуализация мотивов ДНК.
Я не утверждаю, что эти методы оптимальны. Я всё ещё учусь, и это лишь моё представление о решении задачи. Уверена, что есть решения элегантнее.
1. Синтез данных ДНК
Обычно учёные хотят прогнозировать балл связывания, силу экспрессии генов, классифицировать события связывания фактора транскрипции и тому подобное. У нас всё проще. В этом руководстве мы пытаемся научить модель обнаруживать в последовательности ДНК очень маленький и простой паттерн и присваивать ему соответствующий балл. Для этого мы используем глубокое обучение. Но практическая задача даже не в этом. Хочется просто убедиться, что правильно настроили элементы PyTorch и модель может учиться на входных данных, похожих на последовательность ДНК).
Допустим, у нас есть октамерная последовательность ДНК. Начислим ей определённое число баллов за каждый нуклеотид:
- A (аденин) = +20 баллов.
- C (цитозин) = +17 баллов.
- G (гуанин) = +14 баллов.
- T (тимин) = +11 баллов.
Для каждого октамера суммируем балл и усредняем его. Например:
AAAAAAAA даёт нам 20,0
mean(20 + 20 + 20 + 20 + 20 + 20 + 20 + 20) = 20,0
ACAAAAAA даёт 19,625
mean(20 + 17 + 20 + 20 + 20 + 20 + 20 + 20) = 19,625
Баллы выбраны для нуклеотидов произвольно — никакой настоящей биологии в этом нет! Мы просто назначаем последовательностям баллы, чтобы попрактиковаться в работе с PyTorch.
Во многих работах свёрточные нейросети (CNN) определяют «мотивы» или короткие паттерны ДНК. Они могут активировать или подавлять биологические реакции. Поэтому добавим в систему начисления баллов ещё один элемент: иногда нам нужно моделировать мотивы, влияющие на экспрессию генов. Допустим, последовательность получает +10 баллов при наличии мотива TAT в любой части октамера или -10 баллов при наличии мотива GCG. Повторюсь, к реальности это никакого отношения не имеет. Я просто показала вам на примере, как работает простой механизм моделирования активации и подавления.

Простая система подсчёта баллов для октамерных последовательностей ДНК
Вот реализация этой простой системы:
def kmers(k):
'''Generate a list of all k-mers for a given k'''
return [''.join(x) for x in product(['A','C','G','T'], repeat=k)]
seqs8 = kmers(8)
print('Total 8mers:',len(seqs8))
# prints: Total 8mers: 65536
score_dict = {
'A':20,
'C':17,
'G':14,
'T':11
}
def score_seqs_motif(seqs):
'''
Calculate the scores for a list of sequences based on
the above score_dict
'''
data = []
for seq in seqs:
# get the average score by nucleotide
score = np.mean([score_dict[base] for base in seq])
# give a + or - bump if this k-mer has a specific motif
if 'TAT' in seq:
score += 10
if 'GCG' in seq:
score -= 10
data.append([seq,score])
df = pd.DataFrame(data, columns=['seq','score'])
return df
mer8 = score_seqs_motif(seqs8)
Строим распределение баллов для октамерных последовательностей и видим, что они разделились на 3 группы:
- последовательности с мотивом
GCG(~5 баллов); - последовательности без мотивов (~15 баллов);
- последовательности с мотивом
TAT(~25 баллов).

Распределение баллов для октамерных последовательностей
Теперь нам предстоит научить модель прогнозировать балл по последовательности ДНК.
2. Подготовка данных для обучения в PyTorch
Чтобы научить нейросеть прогнозировать балл, нужно дать ей входные данные в виде матрицы чисел. Поясню на примере. Пусть мы хотим разделить фотографии с кошками и без кошек на два набора. Сеть распознаёт изображение как матрицу значений пикселей и изучает паттерны, связанные с относительным расположением пикселей (например, паттерны, соответствующие кошачьим ушам или носу с усами).
Аналогичным образом в матрицу чисел нужно превратить последовательности ДНК (строки ACGT). Но как же нам сделать из нашей ДНК «кошку»?
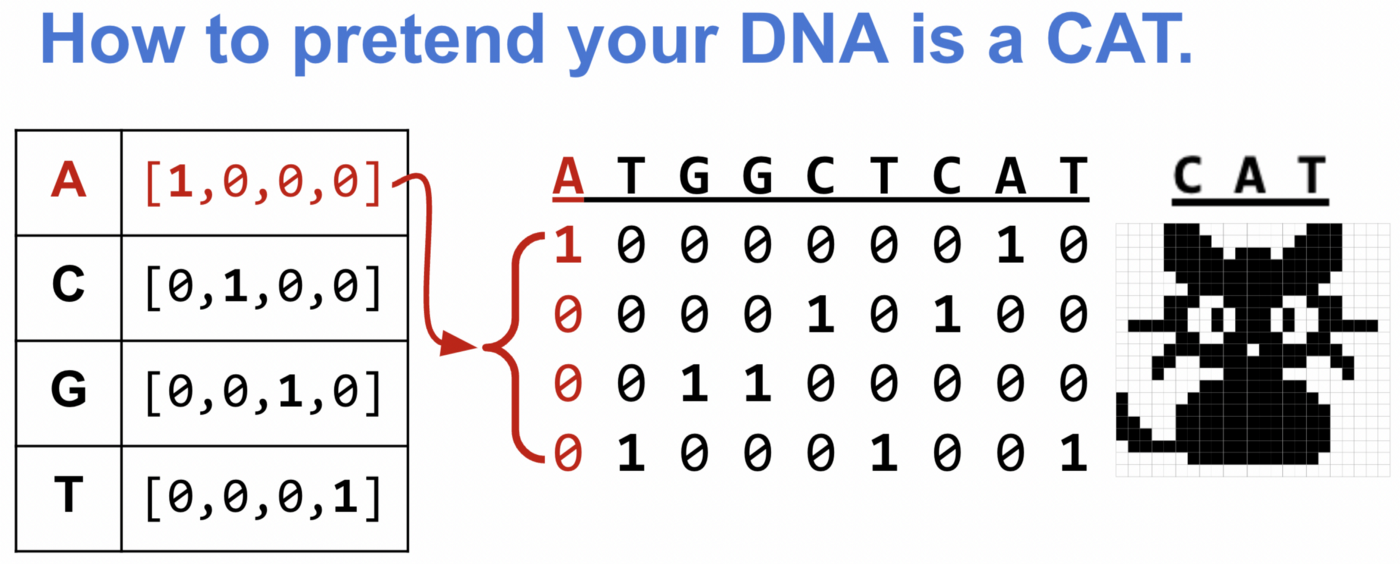
Одна из распространённых стратегий — прямое кодирование [one-hot encoding] ДНК с одним активным состоянием. Для такого кодирования рассмотрим каждый нуклеотид как вектор длиной 4, где в трёх позициях стоят нули, а в одной — единица. Место единицы зависит от типа нуклеотида.
Благодаря прямому кодированию компьютер определит ДНК так же, как кошку на картинке!
def one_hot_encode(seq):
"""
Given a DNA sequence, return its one-hot encoding
"""
# Make sure seq has only allowed bases
allowed = set("ACTGN")
if not set(seq).issubset(allowed):
invalid = set(seq) - allowed
raise ValueError(f"Sequence contains chars not in allowed DNA alphabet (ACGTN): {invalid}")
# Dictionary returning one-hot encoding for each nucleotide
nuc_d = {'A':[1.0,0.0,0.0,0.0],
'C':[0.0,1.0,0.0,0.0],
'G':[0.0,0.0,1.0,0.0],
'T':[0.0,0.0,0.0,1.0],
'N':[0.0,0.0,0.0,0.0]}
# Create array from nucleotide sequence
vec=np.array([nuc_d[x] for x in seq])
return vec
# look at DNA seq of 8 As
a8 = one_hot_encode("AAAAAAAA")
print("AAAAAAAA:\n",a8)
# prints:
# AAAAAAAA:
# [[1. 0. 0. 0.]
# [1. 0. 0. 0.]
# [1. 0. 0. 0.]
# [1. 0. 0. 0.]
# [1. 0. 0. 0.]
# [1. 0. 0. 0.]
# [1. 0. 0. 0.]
# [1. 0. 0. 0.]]
# look at DNA seq of random nucleotides
s = one_hot_encode("AGGTACCT")
print("AGGTACCT:\n",s)
print("shape:",s.shape)
# prints:
# AGGTACCT:
# [[1. 0. 0. 0.]
# [0. 0. 1. 0.]
# [0. 0. 1. 0.]
# [0. 0. 0. 1.]
# [1. 0. 0. 0.]
# [0. 1. 0. 0.]
# [0. 1. 0. 0.]
# [0. 0. 0. 1.]]
# shape: (8, 4)
При помощи прямого кодирования можно подготовить наборы train, val и test. Соответствующая функция quick_split случайным образом выбирает индексы в кадре данных pandas для разделения (функция для этого есть и в sklearn).
В реальных задачах, не связанных с синтезом, может потребоваться более интеллектуальная стратегия разделения. Всё зависит от вашей прогностической задачи. В научных статьях разделение train/test часто производится по хромосоме или по другим особенностям расположения генома.
def quick_split(df, split_frac=0.8):
'''
Given a df of samples, randomly split indices between
train and test at the desired fraction
'''
cols = df.columns # original columns, use to clean up reindexed cols
df = df.reset_index()
# shuffle indices
idxs = list(range(df.shape[0]))
random.shuffle(idxs)
# split shuffled index list by split_frac
split = int(len(idxs)*split_frac)
train_idxs = idxs[:split]
test_idxs = idxs[split:]
# split dfs and return
train_df = df[df.index.isin(train_idxs)]
test_df = df[df.index.isin(test_idxs)]
return train_df[cols], test_df[cols]
full_train_df, test_df = quick_split(mer8)
train_df, val_df = quick_split(full_train_df)
print("Train:", train_df.shape)
print("Val:", val_df.shape)
print("Test:", test_df.shape)
# prints:
# Train: (41942, 2)
# Val: (10486, 2)
# Test: (13108, 2)
При подготовке данных в PyTorch важным шагом является применение объектов DataLoader и Dataset. Чтобы разобраться в этом, мне пришлось изрядно погуглить, а чтобы найти решение, пришлось прочесать кипу документов и постов со StackOverflow!
Dataset заключает данные в объект, которому можно легко задать отформатированные примеры X и маркировку Y для обучаемой модели. DataLoader получает Dataset и ряд других деталей о том, как объединять данные в серии и упрощает прохождение итераций обучения.
from torch.utils.data import Dataset, DataLoader
## Here is a custom defined Dataset object specialized for one-hot encoded DNA:
class SeqDatasetOHE(Dataset):
'''
Dataset for one-hot-encoded sequences
'''
def __init__(self,
df,
seq_col='seq',
target_col='score'
):
# +--------------------+
# | Get the X examples |
# +--------------------+
# extract the DNA from the appropriate column in the df
self.seqs = list(df[seq_col].values)
self.seq_len = len(self.seqs[0])
# one-hot encode sequences, then stack in a torch tensor
self.ohe_seqs = torch.stack([torch.tensor(one_hot_encode(x)) for x in self.seqs])
# +------------------+
# | Get the Y labels |
# +------------------+
self.labels = torch.tensor(list(df[target_col].values)).unsqueeze(1)
def __len__(self): return len(self.seqs)
def __getitem__(self,idx):
# Given an index, return a tuple of an X with it's associated Y
# This is called inside DataLoader
seq = self.ohe_seqs[idx]
label = self.labels[idx]
return seq, label
## Here is how I constructed DataLoaders from Datasets.
def build_dataloaders(train_df,
test_df,
seq_col='seq',
target_col='score',
batch_size=128,
shuffle=True
):
'''
Given a train and test df with some batch construction
details, put them into custom SeqDatasetOHE() objects.
Give the Datasets to the DataLoaders and return.
'''
# create Datasets
train_ds = SeqDatasetOHE(train_df,seq_col=seq_col,target_col=target_col)
test_ds = SeqDatasetOHE(test_df,seq_col=seq_col,target_col=target_col)
# Put DataSets into DataLoaders
train_dl = DataLoader(train_ds, batch_size=batch_size, shuffle=shuffle)
test_dl = DataLoader(test_ds, batch_size=batch_size)
return train_dl,test_dl
train_dl, val_dl = build_dataloaders(train_df, val_df)
Теперь наши объекты DataLoader можно использовать в обучающем цикле!
3. Определение модели PyTorch
Сначала мне хотелось попробовать метод свёрточных нейросетей, потому что в изучении данных генома он показал себя отлично. Для сравнения я взяла простую линейную модель. Вот несколько определений модели:
# very simple linear model
class DNA_Linear(nn.Module):
def __init__(self, seq_len):
super().__init__()
self.seq_len = seq_len
# the 4 is for our one-hot encoded vector length 4!
self.lin = nn.Linear(4*seq_len, 1)
def forward(self, xb):
# reshape to flatten sequence dimension
xb = xb.view(xb.shape[0],self.seq_len*4)
# Linear wraps up the weights/bias dot product operations
out = self.lin(xb)
return out
# basic CNN model
class DNA_CNN(nn.Module):
def __init__(self,
seq_len,
num_filters=32,
kernel_size=3):
super().__init__()
self.seq_len = seq_len
self.conv_net = nn.Sequential(
# 4 is for the 4 nucleotides
nn.Conv1d(4, num_filters, kernel_size=kernel_size),
nn.ReLU(inplace=True),
nn.Flatten(),
nn.Linear(num_filters*(seq_len-kernel_size+1), 1)
)
def forward(self, xb):
# permute to put channel in correct order
# (batch_size x 4channel x seq_len)
xb = xb.permute(0,2,1)
#print(xb.shape)
out = self.conv_net(xb)
return out
Это не оптимальные модели, но надо же с чего-то начинать (ещё раз, мы просто учимся применять PyTorch в контексте ДНК).
- Линейная модель пытается прогнозировать балл по весу нуклеотидов на каждой позиции.
- Модель свёрточных нейросетей использует 32 фильтра с длиной (
kernel_size) 3 для сканирования всей октамерной последовательности и поиска в ней паттернов-тримеров.
4. Определение функций обучающего цикла
Теперь нужно определить цикл обучения. Скажу честно, я не слишком-то уверена в этом решении. Я долго продиралась через ошибки несоответствия размерности матрицы. Наверное, есть красивые подходы! Но, может быть, и так сойдёт? Напишите мне пару слов, если у вас есть свои мысли на этот счёт.
Так или иначе, я пришла к такому определению стека функций:
# adds default optimizer and loss function
run_model()
# loops through epochs
fit()
# loop through batches
train_step()
# calc train loss for batch
loss_batch()
val_step()
# calc val loss for batch
loss_batch()
# +--------------------------------+
# | Training and fitting functions |
# +--------------------------------+
def loss_batch(model, loss_func, xb, yb, opt=None,verbose=False):
'''
Apply loss function to a batch of inputs. If no optimizer
is provided, skip the back prop step.
'''
if verbose:
print('loss batch ****')
print("xb shape:",xb.shape)
print("yb shape:",yb.shape)
print("yb shape:",yb.squeeze(1).shape)
#print("yb",yb)
# get the batch output from the model given your input batch
# ** This is the model's prediction for the y labels! **
xb_out = model(xb.float())
if verbose:
print("model out pre loss", xb_out.shape)
#print('xb_out', xb_out)
print("xb_out:",xb_out.shape)
print("yb:",yb.shape)
print("yb.long:",yb.long().shape)
loss = loss_func(xb_out, yb.float()) # for MSE/regression
# __FOOTNOTE 2__
if opt is not None: # if opt
loss.backward()
opt.step()
opt.zero_grad()
return loss.item(), len(xb)
def train_step(model, train_dl, loss_func, device, opt):
'''
Execute 1 set of batched training within an epoch
'''
# Set model to Training mode
model.train()
tl = [] # train losses
ns = [] # batch sizes, n
# loop through train DataLoader
for xb, yb in train_dl:
# put on GPU
xb, yb = xb.to(device),yb.to(device)
# provide opt so backprop happens
t, n = loss_batch(model, loss_func, xb, yb, opt=opt)
# collect train loss and batch sizes
tl.append(t)
ns.append(n)
# average the losses over all batches
train_loss = np.sum(np.multiply(tl, ns)) / np.sum(ns)
return train_loss
def val_step(model, val_dl, loss_func, device):
'''
Execute 1 set of batched validation within an epoch
'''
# Set model to Evaluation mode
model.eval()
with torch.no_grad():
vl = [] # val losses
ns = [] # batch sizes, n
# loop through validation DataLoader
for xb, yb in val_dl:
# put on GPU
xb, yb = xb.to(device),yb.to(device)
# Do NOT provide opt here, so backprop does not happen
v, n = loss_batch(model, loss_func, xb, yb)
# collect val loss and batch sizes
vl.append(v)
ns.append(n)
# average the losses over all batches
val_loss = np.sum(np.multiply(vl, ns)) / np.sum(ns)
return val_loss
def fit(epochs, model, loss_func, opt, train_dl, val_dl,device,patience=1000):
'''
Fit the model params to the training data, eval on unseen data.
Loop for a number of epochs and keep train of train and val losses
along the way
'''
# keep track of losses
train_losses = []
val_losses = []
# loop through epochs
for epoch in range(epochs):
# take a training step
train_loss = train_step(model,train_dl,loss_func,device,opt)
train_losses.append(train_loss)
# take a validation step
val_loss = val_step(model,val_dl,loss_func,device)
val_losses.append(val_loss)
print(f"E{epoch} | train loss: {train_loss:.3f} | val loss: {val_loss:.3f}")
return train_losses, val_losses
def run_model(train_dl,val_dl,model,device,
lr=0.01, epochs=50,
lossf=None,opt=None
):
'''
Given train and val DataLoaders and a NN model, fit the mode to the training
data. By default, use MSE loss and an SGD optimizer
'''
# define optimizer
if opt:
optimizer = opt
else: # if no opt provided, just use SGD
optimizer = torch.optim.SGD(model.parameters(), lr=lr)
# define loss function
if lossf:
loss_func = lossf
else: # if no loss function provided, just use MSE
loss_func = torch.nn.MSELoss()
# run the training loop
train_losses, val_losses = fit(
epochs,
model,
loss_func,
optimizer,
train_dl,
val_dl,
device)
return train_losses, val_losses
5. Запуск моделей
Для начала испытаем на наших октамерных последовательностях линейный подход.
# use GPU if available
DEVICE = torch.device('cuda' if torch.cuda.is_available() else 'cpu')
# get the sequence length from the first seq in the df
seq_len = len(train_df['seq'].values[0])
# create Linear model object
model_lin = DNA_Linear(seq_len)
model_lin.to(DEVICE) # put on GPU
# run the model with default settings!
lin_train_losses, lin_val_losses = run_model(
train_dl,
val_dl,
model_lin,
DEVICE
)
Соберём данные о потерях train и val и набросаем график:
def quick_loss_plot(data_label_list,loss_type="MSE Loss",sparse_n=0):
'''
For each train/test loss trajectory, plot loss by epoch
'''
for i,(train_data,test_data,label) in enumerate(data_label_list):
plt.plot(train_data,linestyle='--',color=f"C{i}", label=f"{label} Train")
plt.plot(test_data,color=f"C{i}", label=f"{label} Val",linewidth=3.0)
plt.legend()
plt.ylabel(loss_type)
plt.xlabel("Epoch")
plt.legend(bbox_to_anchor=(1,1),loc='upper left')
plt.show()
lin_data_label = (lin_train_losses,lin_val_losses,"Lin")
quick_loss_plot([lin_data_label])

Обучение линейной модели и оценка кривых потерь
На первый взгляд, такой подход привнёс в обучение немного.
Теперь попробуем метод свёрточных нейросетей и построим график для него.
seq_len = len(train_df['seq'].values[0])
# create Linear model object
model_cnn = DNA_CNN(seq_len)
model_cnn.to(DEVICE) # put on GPU
# run the model with default settings!
cnn_train_losses, cnn_val_losses = run_model(
train_dl,
val_dl,
model_cnn,
DEVICE
)
cnn_data_label = (cnn_train_losses,cnn_val_losses,"CNN")
quick_loss_plot([lin_data_label,cnn_data_label])

Кривые потерь для линейного метода и метода свёрточных нейросетей (CNN)
Очевидно, что CNN может уловить паттерн, а линейный метод не может. Прогоним несколько последовательностей и посмотрим, что получится.
# oracle dict of true score for each seq
oracle = dict(mer8[['seq','score']].values)
def quick_seq_pred(model, desc, seqs, oracle):
'''
Given a model and some sequences, get the model's predictions
for those sequences and compare to the oracle (true) output
'''
print(f"__{desc}__")
for dna in seqs:
s = torch.tensor(one_hot_encode(dna)).unsqueeze(0).to(DEVICE)
pred = model(s.float())
actual = oracle[dna]
diff = pred.item() - actual
print(f"{dna}: pred:{pred.item():.3f} actual:{actual:.3f} ({diff:.3f})")
def quick_8mer_pred(model, oracle):
seqs1 = ("poly-X seqs",['AAAAAAAA', 'CCCCCCCC','GGGGGGGG','TTTTTTTT'])
seqs2 = ("other seqs", ['AACCAACA','CCGGTGAG','GGGTAAGG', 'TTTCGTTT'])
seqsTAT = ("with TAT motif", ['TATAAAAA','CCTATCCC','GTATGGGG','TTTATTTT'])
seqsGCG = ("with GCG motif", ['AAGCGAAA','CGCGCCCC','GGGCGGGG','TTGCGTTT'])
TATGCG = ("both TAT and GCG",['ATATGCGA','TGCGTATT'])
for desc,seqs in [seqs1, seqs2, seqsTAT, seqsGCG, TATGCG]:
quick_seq_pred(model, desc, seqs, oracle)
print()
# Ask the trained Linear model to make
# predictions for some 8-mers
quick_8mer_pred(model_lin, oracle)
# prints:
# __poly-X seqs__
# AAAAAAAA: pred:23.230 actual:20.000 (3.230)
# CCCCCCCC: pred:13.582 actual:17.000 (-3.418)
# GGGGGGGG: pred:7.006 actual:14.000 (-6.994)
# TTTTTTTT: pred:17.767 actual:11.000 (6.767)
# __other seqs__
# AACCAACA: pred:18.818 actual:18.875 (-0.057)
# CCGGTGAG: pred:12.205 actual:15.125 (-2.920)
# GGGTAAGG: pred:13.826 actual:15.125 (-1.299)
# TTTCGTTT: pred:14.815 actual:12.125 (2.690)
# __with TAT motif__
# TATAAAAA: pred:22.146 actual:27.750 (-5.604)
# CCTATCCC: pred:16.931 actual:25.875 (-8.944)
# GTATGGGG: pred:12.141 actual:24.000 (-11.859)
# TTTATTTT: pred:18.266 actual:22.125 (-3.859)
# __with GCG motif__
# AAGCGAAA: pred:16.736 actual:8.125 (8.611)
# CGCGCCCC: pred:12.346 actual:6.250 (6.096)
# GGGCGGGG: pred:7.907 actual:4.375 (3.532)
# TTGCGTTT: pred:12.839 actual:2.500 (10.339)
# __both TAT and GCG__
# ATATGCGA: pred:15.664 actual:15.875 (-0.211)
# TGCGTATT: pred:14.771 actual:13.625 (1.146)
Из приведённых выше примеров видно, что линейная модель занижает прогноз для последовательности с большим количеством G и завышает его для последовательности с большим количеством T. По-видимому, она просто не замечает, что последовательности с GCG имеют необычно низкие баллы, а последовательности с TAT — необычно высокие. Но, поскольку в линейной модели невозможно учесть различия между GCG и GAG, она просто понижает балл для последовательности с G. Из нашей схемы подсчёта баллов известно, что это не так. Дело тут не в том, что G вредны или, если хотите, субвитальны, а в том, что вредны именно мотивы GCG.
# Ask the trained CNN model to make
# predictions for some 8-mers
quick_8mer_pred(model_cnn, oracle)
# prints:
# __poly-X seqs__
# AAAAAAAA: pred:19.722 actual:20.000 (-0.278)
# CCCCCCCC: pred:16.620 actual:17.000 (-0.380)
# GGGGGGGG: pred:13.771 actual:14.000 (-0.229)
# TTTTTTTT: pred:10.767 actual:11.000 (-0.233)
# __other seqs__
# AACCAACA: pred:18.530 actual:18.875 (-0.345)
# CCGGTGAG: pred:14.925 actual:15.125 (-0.200)
# GGGTAAGG: pred:14.900 actual:15.125 (-0.225)
# TTTCGTTT: pred:11.789 actual:12.125 (-0.336)
# __with TAT motif__
# TATAAAAA: pred:26.154 actual:27.750 (-1.596)
# CCTATCCC: pred:24.321 actual:25.875 (-1.554)
# GTATGGGG: pred:22.870 actual:24.000 (-1.130)
# TTTATTTT: pred:20.581 actual:22.125 (-1.544)
# __with GCG motif__
# AAGCGAAA: pred:8.689 actual:8.125 (0.564)
# CGCGCCCC: pred:6.531 actual:6.250 (0.281)
# GGGCGGGG: pred:5.029 actual:4.375 (0.654)
# TTGCGTTT: pred:3.055 actual:2.500 (0.555)
# __both TAT and GCG__
# ATATGCGA: pred:15.178 actual:15.875 (-0.697)
# TGCGTATT: pred:12.853 actual:13.625 (-0.772)
CNN лучше улавливает разницу между тримерными мотивами! При этом он хорошо переваривает последовательности и с мотивами и без них.
6. Проверка прогнозов модели на тестовом наборе данных
В любой задаче машинного обучения при оценке результатов важно убедиться, что модель может делать хорошие прогнозы по тестовым наборам, с которыми она никогда не сталкивалась в ходе обучения. Разница между реальными тестовыми последовательностями и прогнозами модели лучше всего видна на сравнительных графиках.
import altair as alt
from sklearn.metrics import r2_score
def parity_plot(model_name,df,r2):
'''
Given a dataframe of samples with their true and predicted values,
make a scatterplot.
'''
plt.scatter(df['truth'].values, df['pred'].values, alpha=0.2)
# y=x line
xpoints = ypoints = plt.xlim()
plt.plot(xpoints, ypoints, linestyle='--', color='k', lw=2, scalex=False, scaley=False)
plt.ylim(xpoints)
plt.ylabel("Predicted Score",fontsize=14)
plt.xlabel("Actual Score",fontsize=14)
plt.title(f"{model_name} (r2:{r2:.3f})",fontsize=20)
plt.show()
def alt_parity_plot(model,df, r2):
'''
Make an interactive parity plot with altair
'''
chart = alt.Chart(df).mark_circle(size=100,opacity=0.4).encode(
alt.X('truth:Q'),
alt.Y('pred:Q'),
tooltip=['seq:N']
).properties(
title=f'{model} (r2:{r2:.3f})'
).interactive()
chart.save(f'alt_out/parity_plot_{model}.html')
display(chart)
def parity_pred(models, seqs, oracle,alt=False):
'''Given some sequences, get the model's predictions '''
dfs = {} # key: model name, value: parity_df
for model_name,model in models:
print(f"Running {model_name}")
data = []
for dna in seqs:
s = torch.tensor(one_hot_encode(dna)).unsqueeze(0).to(DEVICE)
actual = oracle[dna]
pred = model(s.float())
data.append([dna,actual,pred.item()])
df = pd.DataFrame(data, columns=['seq','truth','pred'])
r2 = r2_score(df['truth'],df['pred'])
dfs[model_name] = (r2,df)
#plot parity plot
if alt: # make an altair plot
alt_parity_plot(model_name, df, r2)
else:
parity_plot(model_name, df, r2)
# generate plots
seqs = test_df['seq'].values
models = [
("Linear", model_lin),
("CNN", model_cnn)
]
parity_pred(models, seqs, oracle)

Сравнение графиков показывает, насколько точно модель прогнозирует отдельные последовательности. У идеальной модели все графики точно лягут на диагональ y=x. Это покажет, что модель точно прогнозирует последовательности. Отклонения от диагонали y=x означают, что прогнозы модели завышены или занижены.
Как мы видим, линейная модель может в какой-то мере прогнозировать линию тренда тестовых последовательностей, но обилие последовательностей в нижней и верхней областях распределения (при наличии мотивов) легко сбивают эту модель с толку.
CNN работает гораздо лучше и ожидаемо предсказывает значения, близкие к реальным, ведь архитектура нашего CNN использует ядра тримеров для поиска ключевых мотивов вдоль последовательности.
Впрочем, CNN — тоже не идеал. Можно было бы обучать его дольше или корректировать гиперпараметры, но цель здесь не в совершенстве. Мы сильно упростили задачу по сравнению с реальными регулятивными грамматиками. И я подумала, что для интерактивного выявления последовательностей, с которыми модели не справляются, было бы интересно использовать библиотеку визуализации Altair:
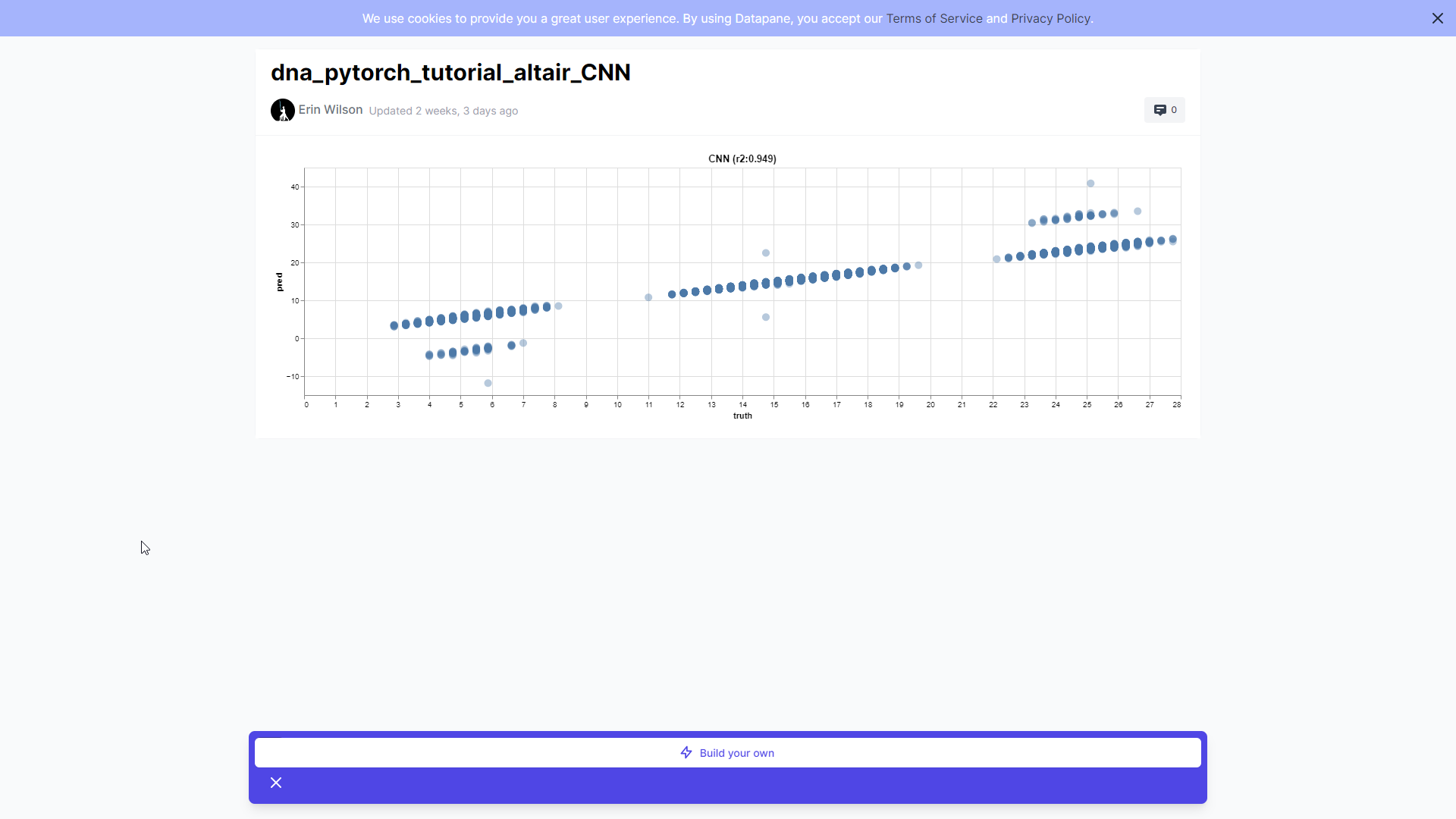
Заметьте, что вне диагонали наши последовательности, как правило, имеют множественные экземпляры мотивов! При подсчёте баллов мы просто давали последовательности ± поправки за один мотив. В действительности же разумно было бы несколько таких «бонусов» за несколько мотивов. В своём примере я просто даю «бонус» за 1 экземпляр мотива, но функцию расчёта баллов можно было бы составить иначе.
В любом случае здорово, когда модель замечает множественные экземпляры и прогнозирует их важность. Кажется, здесь я немного обманула её… Впрочем, R2 0,95 — это уже не так плохо.
7. Визуализация свёрточных фильтров
При обучении моделей CNN полезно визуализировать свёрточные фильтры первого слоя, чтобы лучше понять процесс обучения. При работе с изображениями свёрточные фильтры первого слоя часто изучают паттерны границ, цвета или текстуры — основные элементы изображения, при объединении которых мы получаем более сложные признаки.
В случае ДНК мы исходим из того, что свёрточные фильтры можно рассматривать как сканеры мотивов. По аналогии с матрицей весовых коэффициентов позиций для визуализации логотипов последовательностей, свёрточный фильтр похож на матрицу, показывающую определённый паттерн ДНК. Однако вместо того, чтобы быть точной последовательностью, он может содержать некоторую неопределённость относительно того, какие нуклеотиды показываются в той или иной части паттерна. Некоторые позиции могут быть строго определены (например, в позиции 2 всегда есть A; информативность высокая), в других же с почти равной вероятностью может быть множество нуклеотидов (энтропия высокая, информативность низкая).
Вычисления на скрытых слоях нейросетей могут быть очень сложными, и не каждый свёрточный фильтр будет представлять собой очевидный паттерн. Однако фильтры всё же выявляют информативные для объяснения прогноза паттерны.
Ниже приведены некоторые функции для визуализации свёрточных фильтров первого слоя как в виде необработанной тепловой карты, так и в виде логотипа.
import logomaker
def get_conv_layers_from_model(model):
'''
Given a trained model, extract its convolutional layers
'''
model_children = list(model.children())
# counter to keep count of the conv layers
model_weights = [] # we will save the conv layer weights in this list
conv_layers = [] # we will save the actual conv layers in this list
bias_weights = []
counter = 0
# append all the conv layers and their respective weights to the list
for i in range(len(model_children)):
# get model type of Conv1d
if type(model_children[i]) == nn.Conv1d:
counter += 1
model_weights.append(model_children[i].weight)
conv_layers.append(model_children[i])
bias_weights.append(model_children[i].bias)
# also check sequential objects' children for conv1d
elif type(model_children[i]) == nn.Sequential:
for child in model_children[i]:
if type(child) == nn.Conv1d:
counter += 1
model_weights.append(child.weight)
conv_layers.append(child)
bias_weights.append(child.bias)
print(f"Total convolutional layers: {counter}")
return conv_layers, model_weights, bias_weights
def view_filters(model_weights, num_cols=8):
model_weights = model_weights[0]
num_filt = model_weights.shape[0]
filt_width = model_weights[0].shape[1]
num_rows = int(np.ceil(num_filt/num_cols))
# visualize the first conv layer filters
plt.figure(figsize=(20, 17))
for i, filter in enumerate(model_weights):
ax = plt.subplot(num_rows, num_cols, i+1)
ax.imshow(filter.cpu().detach(), cmap='gray')
ax.set_yticks(np.arange(4))
ax.set_yticklabels(['A', 'C', 'G','T'])
ax.set_xticks(np.arange(filt_width))
ax.set_title(f"Filter {i}")
plt.tight_layout()
plt.show()
conv_layers, model_weights, bias_weights = get_conv_layers_from_model(model_cnn)
view_filters(model_weights)

Возможно, это полезная информация, но обычно люди любят визуализировать последовательности с некоторой неопределённостью в виде логотипов мотивов, откладывая позиции мотива по X, а вероятности появления нуклеотидов в каждой из них — по Y. Часто для удобства визуализации эти вероятности преобразуются в биты (информацию).
Для преобразования необработанных свёрточных фильтров в визуальные матрицы весовых коэффициентов позиций обычно собирают активации фильтров: применяют веса фильтра вдоль кодируемой прямо последовательности и измеряют активацию фильтра (а также то, насколько хорошо веса соответствуют последовательности).
Матрицы весовых коэффициентов фильтра, соответствующие близкому совпадению с последовательностью, будут высокоактивированными (то есть дадут более высокие оценки совпадения). Собирая подпоследовательности ДНК, которые дают самые высокие оценки активации, мы можем создать весовую матрицу «высокоактивированных последовательностей» для каждого фильтра и, таким образом, визуализировать свёрточный фильтр в виде логотипа мотива.

Схема, показывающая степень сбора активированных участков последовательности и их преобразования в логотипы мотивов для фильтра свёртки
def get_conv_output_for_seq(seq, conv_layer):
'''
Given an input sequeunce and a convolutional layer,
get the output tensor containing the conv filter
activations along each position in the sequence
'''
# format seq for input to conv layer (OHE, reshape)
seq = torch.tensor(one_hot_encode(seq)).unsqueeze(0).permute(0,2,1).to(DEVICE)
# run seq through conv layer
with torch.no_grad(): # don't want as part of gradient graph
# apply learned filters to input seq
res = conv_layer(seq.float())
return res[0]
def get_filter_activations(seqs, conv_layer,act_thresh=0):
'''
Given a set of input sequences and a trained convolutional layer,
determine the subsequences for which each filter in the conv layer
activate most strongly.
1.) Run seq inputs through conv layer.
2.) Loop through filter activations of the resulting tensor, saving the
position where filter activations were > act_thresh.
3.) Compile a count matrix for each filter by accumulating subsequences which
activate the filter above the threshold act_thresh
'''
# initialize dict of pwms for each filter in the conv layer
# pwm shape: 4 nucleotides X filter width, initialize to 0.0s
num_filters = conv_layer.out_channels
filt_width = conv_layer.kernel_size[0]
filter_pwms = dict((i,torch.zeros(4,filt_width)) for i in range(num_filters))
print("Num filters", num_filters)
print("filt_width", filt_width)
# loop through a set of sequences and collect subseqs where each filter activated
for seq in seqs:
# get a tensor of each conv filter activation along the input seq
res = get_conv_output_for_seq(seq, conv_layer)
# for each filter and it's activation vector
for filt_id,act_vec in enumerate(res):
# collect the indices where the activation level
# was above the threshold
act_idxs = torch.where(act_vec>act_thresh)[0]
activated_positions = [x.item() for x in act_idxs]
# use activated indicies to extract the actual DNA
# subsequences that caused filter to activate
for pos in activated_positions:
subseq = seq[pos:pos+filt_width]
#print("subseq",pos, subseq)
# transpose OHE to match PWM orientation
subseq_tensor = torch.tensor(one_hot_encode(subseq)).T
# add this subseq to the pwm count for this filter
filter_pwms[filt_id] += subseq_tensor
return filter_pwms
def view_filters_and_logos(model_weights,filter_activations, num_cols=8):
'''
Given some convolutional model weights and filter activation PWMs,
visualize the heatmap and motif logo pairs in a simple grid
'''
model_weights = model_weights[0].squeeze(1)
print(model_weights.shape)
# make sure the model weights agree with the number of filters
assert(model_weights.shape[0] == len(filter_activations))
num_filts = len(filter_activations)
num_rows = int(np.ceil(num_filts/num_cols))*2+1
# ^ not sure why +1 is needed... complained otherwise
plt.figure(figsize=(20, 17))
j=0 # use to make sure a filter and it's logo end up vertically paired
for i, filter in enumerate(model_weights):
if (i)%num_cols == 0:
j += num_cols
# display raw filter
ax1 = plt.subplot(num_rows, num_cols, i+j+1)
ax1.imshow(filter.cpu().detach(), cmap='gray')
ax1.set_yticks(np.arange(4))
ax1.set_yticklabels(['A', 'C', 'G','T'])
ax1.set_xticks(np.arange(model_weights.shape[2]))
ax1.set_title(f"Filter {i}")
# display sequence logo
ax2 = plt.subplot(num_rows, num_cols, i+j+1+num_cols)
filt_df = pd.DataFrame(filter_activations[i].T.numpy(),columns=['A','C','G','T'])
filt_df_info = logomaker.transform_matrix(filt_df,from_type='counts',to_type='information')
logo = logomaker.Logo(filt_df_info,ax=ax2)
ax2.set_ylim(0,2)
ax2.set_title(f"Filter {i}")
plt.tight_layout()
# just use some seqs from test_df to activate filters
some_seqs = random.choices(seqs, k=3000)
filter_activations = get_filter_activations(some_seqs, conv_layers[0],act_thresh=1)
view_filters_and_logos(model_weights,filter_activations)
# prints:
# Num filters 32
# filt_width 3
# torch.Size([32, 4, 3])

Из конкретно этого обучения CNN видно, что несколько фильтров уловили сильные мотивы TAT и GCG, в то время как другие фильтры сфокусировались и на других паттернах.
О важности визуализации свёрточных фильтров для интерпретации модели нет единого мнения. В глубоких моделях с несколькими свёрточными слоями свёрточные фильтры могут комбинироваться в скрытых слоях более сложными способами, поэтому фильтры первого слоя сами по себе могут быть не такими информативными (Ку и Эдди, 2019]). Большая часть этой области с тех пор перешла к механизмам внимания и другим методам объяснения результатов. Однако, если вам интересно визуализировать фильтры как потенциальные мотивы, эти функции могут быть полезны!
8. Итоги
В руководстве даются основы построения моделей CNN в PyTorch для обработки последовательностей ДНК. Практическая часть работы не отражает реалии биологических сигналов. Наш метод подсчёта баллов, скорее, моделирует наличие регуляторных мотивов в очень коротких последовательностях, которые легко понять человеку. Поведение PyTorch соответствовало ожиданиям. В этом маленьком примере мы проверили способность базовой CNN со сдвиговыми фильтрами к прогнозу расчёта баллов. CNN показала себя лучше, чем линейный метод, выявивший только абсолютные положения нуклеотидов (без учёта местных особенностей).
Подробнее о применении CNN к ДНК в естественных условиях можно узнать из следующих работ:
- DeepBind: Alipanahi et al 2015.
- DeepSea: Zhou and Troyanskaya 2015.
- Basset: Kelley et al 2016.
Надеюсь, эта статья поможет с PyTorch всем, кто интересуется биологией и ДНК, но мало знаком с машинным обучением.
9. Примечания
Первое
В нашем примере свёрточной нейросети используется одномерный свёрточный слой. Поскольку ДНК — не двухмерная картинка, достаточно прогнать Conv1D по всей длине последовательности, не сканируя «вниз» и «вверх». (Сдвиг фильтра «вверх» и «вниз» неприменим при кодировании матриц ДНК с одним активным состоянием. Нет смысла отделять ряды A и C от G и T — все четыре ряда должны быть строго представлены в последовательности).
Однако мне однажды понадобился инструмент анализа от keras, и тогда я нашла скрипт преобразования pytorch2keras. Скрипт преобразования умеет работать только со слоями Conv2d и выдаёт ошибки по слоям Conv1d в модели.
Если он понадобится и вам, вот пример переформатирования CNN в Conv2D, который обеспечивает беспроблемное сканирование, как для Conv1D:
class DNA_CNN_2D(nn.Module):
def __init__(self,
seq_len,
num_filters=31,
kernel_size=3,
):
super().__init__()
self.seq_len = seq_len
self.conv_net = nn.Sequential(
nn.Conv2d(1, num_filters, kernel_size=(4,kernel_size)),
# ^^ changed from 4 to 1 channel, moved 4 to kernel_size
nn.ReLU(),
nn.Flatten(),
nn.Linear(num_filters*(seq_len-kernel_size+1), 1)
)
def forward(self, xb):
# reshape view to batch_ssize x 4channel x seq_len
# permute to put channel in correct order
xb = xb.permute(0,2,1).unsqueeze(1)
# ^^ Conv2D input fix
#print(xb.shape)
out = self.conv_net(xb)
return out
Второе
Если ваша задача связана с классификацией, а не с регрессией, возможно, вы предпочтёте использовать CrossEntropyLoss. Учтите, что CrossEntropyLoss требуется формат, отличный от MSELoss. Попробуйте такой вариант:
loss = loss_func(xb_out, yb.long().squeeze (1))
А пока учёные моделируют ДНК, мы научим вас разрабатывать системы ИИ, которые решают широкий круг проблем:Чтобы увидеть все курсы, кликните по баннеру:

- Профессия Data Scientist
- Профессия Data Analyst
- Курс «Математика для Data Science»
- Курс «Математика и Machine Learning для Data Science»
- Курс по Data Engineering
- Курс «Machine Learning и Deep Learning»
- Курс по Machine Learning
Python, веб-разработка
- Профессия Fullstack-разработчик на Python
- Курс «Python для веб-разработки»
- Профессия Frontend-разработчик
- Профессия Веб-разработчик
Мобильная разработка
Java и C#
- Профессия Java-разработчик
- Профессия QA-инженер на JAVA
- Профессия C#-разработчик
- Профессия Разработчик игр на Unity
От основ — в глубину
А также