
Эффект химического вещества в качестве лекарства проявляется, когда оно связывается с целевым белком. В этом случае вещество меняет свою форму в соответствии с формой целевого белка. Уровень изменения напрямую связан с аффинностью* связывания вещества и белка и позволяет получить общее представление об эффективности лекарства. В процессе исследований ученым очень важно точно спрогнозировать этот показатель.
Для подсчета уровня изменения химического вещества используются методики, основанные на принципах квантовой или ньютоновской механики. Неэмперические расчеты на основе квантовой механики обеспечивают высочайшую точность, представляя собой анализ состояния электронов на основе типа и положения атомов. Однако выполнение подобных исследований занимает очень много времени. Для точного моделирования уровня изменения химических веществ с помощью этого метода требуются годы, что делает его не пригодным для практического применения.
Напротив, приближенные вычисления на основе молекулярного моделирования выполняются очень быстро и широко распространены в науке. Они используют принципы ньютоновской механики для подсчета силы взаимодействия между атомами и могут применяться для выявления состояния больших молекул, включая белок.
С точки зрения ньютоновской механики, силы, возникающие между атомами, выражаются следующим образом:
- Как сила, которая зависит от расстояния между двумя связанными атомами;
- Как сила, которая зависит от углов между тремя связанными атомами;
- Как сила, которая зависит от уровня скрученности в связке;
- Как сила, которая зависит от расстояния между несвязанными атомами.
Когда химическое вещество связано с целевым белком, уровень скрученности связки отражает степень деформации. Тем не менее, при использовании существующих технологий точность определения двугранного угла, значение которого необходимо для подсчета скрученности связки, достаточно низкая, что вызывает проблему низкой точности определения аффинности связывания.
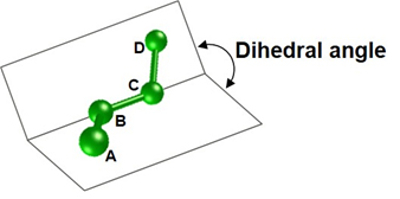
Двугранный угол (угол, образованный плоскостью атомов A, B и C и плоскостью атомов B, C и D)
В чем суть разработки Fujitsu?
Fujitsu занимается разработкой молекулярного моделирования более десяти лет. Используя накопленный опыт, ученые компании создали технологию, которая учитывает не только места примыкания, в которых возникает скрученность, но и влияние расположенных рядом атомов друг на друга.
Существующая технология оценивает значение двухгранных углов на основании положения от 4 до 2 атомов в связке и других атомов, к которым привязаны эти атомы. Однако, в зависимости от структуры молекулы, существуют случаи, когда атомы, не включенные в эти 4 учитываемых атома, оказывают большое влияние на соседние, и в таких случаях высока вероятность возникновения ошибок в расчетах.
Специалисты Fujitsu создали базу данных формул оценки для тех случаев, когда влияние атомов, расположенных далеко от связки, принципиально важно. Используя соответствующую формулу оценки, теперь можно выполнять точную оценку молекулярной скрученности, чего ранее сделать было нельзя.

Пример молекулярной структуры: 3-(метиламино) пиразол
После того как Fujitsu интегрировала эту технологию со специализированным ПО, в лабораторных условиях была изучена точность вычислений новой разработки. Fujitsu провела оценку этой технологии для 190 типов химических веществ, сравнивая полученные результаты с верными, полученными в ходе неэмпирических расчетов, и подсчитывая коэффициент погрешности. Исследование позволило доказать, что вероятность возникновения ошибок при оценке уровня скрученности у новой разработки, в среднем, в 10 раз ниже по сравнению с используемой ранее технологией.
Практическое применение

Оценка параметра двугранного угла с использованием 190 типов структур химических веществ
Новый способ определения аффинности связывания целевых белков и химических веществ демонстрирует гораздо более точные результаты измерений по сравнению с ранее используемой технологией General AMBER Force Field 1.8 (GAFF 1.8). Ученые предполагают, что ее практическое внедрение позволит создать принципиально новые лекарственные средства. Компания Fujitsu также планирует включить эту технологию в собственный сервис разработки лекарств.
*термодинамическая характеристика, количественно описывающая силу взаимодействия веществ
Комментарии (24)

mickvav
18.06.2018 15:35А можно чуть подробнее — ссылки на статьи там, или это чисто внутренние разработки и никто ничего не публикует?
Fujitsu_Admin
18.06.2018 16:02На данный момент это внутренняя разработка компании, которую в будущем планируется использовать в рамках собственного специализированного сервиса.
mickvav
18.06.2018 16:57И как вы тогда собрались убеждать ученых, работающих с открытыми инструментами (gromacs, например), что ваш внутренний сервис работает лучше, чем существующие модели (коих несколько и разных) если не собираетесь публиковаться в peer-reviewed журналах? Маркетинговыми статьями на хабре? Seriously?
BalinTomsk
18.06.2018 18:06То что люди открывают некоторые стороны своих разработок и показывают куда и как двигаются — это уже большой плюс.
Не все что бесплатное работает лучше во всем чем платное.KonkovVladimir
19.06.2018 07:50Наиболее правильным способом нахождения наблюдаемых свойств молекул является решение (операторного) уравнения Шрёдингера и нахождение его собственных значений и векторов, т.е. волновой функции из которой затем можно получить значение любой наблюдаемой в эксперименте величины.
Поскольку молекула является квантовой системой и для каждой молекулы существует мапинги на соответствующие квантовые компьютеры вычисляющие ее (методы Bravyi-Kitaev и Jordan-Wigner arxiv.org/pdf/1712.00446.pdf), то в общем случае решение задачи экспонециально сложно.
Однако если атомы заменить шариками, а взаимодействия между ними представить в видепружинокНьютоновских сил (О-о-о!) вычисляемых через набор подгоночных параметров, то можно подогнать эти параметры под заранее выбранный датасет.
Куда двигаются люди понятно — увеличивают количество подгоночных параметров, расширяют датасет.
DaylightIsBurning
18.06.2018 18:43Вангую, что если сами не откроют, то через 2-4 года кто-то из научных групп разработает открытый аналог. Идея-то простая, вся инфраструктура есть.
KonkovVladimir
18.06.2018 17:53В по какой методике проводилось сравнение с General AMBER Force Field 1.8 (GAFF 1.8)? Достаточно ссылки на статью (или DOI).
А то вспоминаются «забаненые в Gaussian» www.bannedbygaussian.org
Gaussian одна из первых программ для расчетов по квантовой химии, была разработана в начале 70-х, соглашаясь с лицензионным соглашением пользователь не имеет права публично сравнивать производительность Gaussian с аналогичными программами. Нарушители отправлялись в бан целыми университетами и был даже один нобелевский лауреат
www.nobelprize.org/nobel_prizes/chemistry/laureates/1998/pople-bio.html
SeLarin
20.06.2018 08:56Эммм… Fujitsu открыли силовые поля второго рода и перекрёстные члены? Так это не ново.
DaylightIsBurning
20.06.2018 19:27+1Ново то, что они добавили зависимость силовых констант двугранного члена силового поля от «несвязанных» углов/расстояний. По принципу похоже на improper torsions и polarizable force fields.
arielf
21.06.2018 01:40Мысль ясна в целом и не нова — заменить «честные» квантово физические расчёты (причём в квантовой механике и так немало приближений) на классико механические. Перевести сложность из экспоненциальной в полиномиальную. Но всё равно степени полиномов как правило большие, и точность не очень хорошая, ибо нужно много подгоночных параметров. Но как не подгоняй — всё равно что-либо упустишь. В общем, в таких расчётах — называемых ab initio — нужны реальные квантовые компы.
DaylightIsBurning
21.06.2018 13:54Такие квантовые компы неизвестно когда будут, а когда будут, то неизвестно, насколько полезны будут для решения задач молекулярной динамики. Тем временем, метод «классической» молекулярной механики худо-бедно работает и с пользой применяется уже сейчас.
arielf
21.06.2018 15:12Ну как бы моделирование квантовых процессов — «нативная» проблема квантовых компов (как и разложение на простые числа), которую они решают за полиномиальное время. Вы бы глянули про квантовые вычисления вначале.
DaylightIsBurning
21.06.2018 15:18Я в курсе, но вы теоретезируете, а каковы будут возможности и ограничения квантовых компьютеров в контексте задачи молекулярной динамики / квантовой химии не знает на данный момент никто. Сроки появления и стоимость также неизвестны. А возможности молекулярной динамики на доступных сегодня компьютерах ясны и достаточны для извлечения пользы сегодня.
Утверждение «в ab initio расчётах нужны реальные квантовые компы» подобно высказыванию «для полётов в космос нужны технологии антигравитации».
Desavian
очень круто, правда до практической реализации как до луны пешком, но то, что в этом направлении движутся — сильно радует, лет через 15-20 возможно начнут использовать при разработке лекарств и сроки внедрения сократятся раз в пять…
главное чтобы на всю отрасль компьютерного моделирования перестали смотреть как на упоровшихся фриков и начали активно использовать их наработки, как только хоть что-то взлетит и поломает текущую структуру внедрения, весь механизм изменится в лучшую сторону.
vesper-bot
Мощностей на такие штуки надо много. Помнится, был на BOINC проект POEM, считал помимо прочего и взаимодействие белков с какими-то соединениями. Жрал как я не знаю что, 16 часов на одну подзадачу (комп правда не очень мощный), а подзадач там на эксперимент было несколько сотен тысяч. Дык, закрылся, что-то серьезное посчитав, а результаты применить никто не может — денег не поиметь, лицензия BOINC не позволяет. Если кто-то затеет за вменяемое время получить подобные результаты, ему придется городить полноценный суперкомпьютер, или где-то арендовать мощности — или идти в мировую науку, но терять профит от разработок. Куда ни кинь, везде потери денег ИМХО колоссальные.
DaylightIsBurning
Сейчас многие вещи считаются на видеокартах, примерно на порядок эффективнее чем 5 лет назад. То, на что в 2012 нужно было 300 CPU*month сегодня делается за менее чем 1 GPU*month.
vesper-bot
ЕМНИП эти 16 часов были на GPU (nVidia GT210) уже, неспроста же у меня 16М камней в нем было. Но вы правы в том, что на ГП считать быстрее.
DaylightIsBurning
Современный nVidia GTX 1080 Ti примерно в 300 раз производительнее nVidia GT210. Плюс за пять лет GPU-код стал раза в два эффективнее. Задача, которая считалась за 300000*16часов на GT210, сегодня посчитается за две недели на двух нодах по 10 GTX 1080 Ti. Научная группа, в которой я работаю располагает примерно 200 штук собственных GTX 1080 Ti + дополнительно время на Jureca под отдельные проекты. В группе ~25 учёных.
adeptoleg
Всё в принципе решаемо и зависит от реализации потенциала. Как пример тот же Folding@home в свое время переплюнул по мощности вычислений парочку суперкомпьютеров того времени. Я сам к примеру в порыве энтузиазма себе ставил их софт. Главное чтоб результат был реально применим.
Alex023
boinc.bakerlab.org/rosetta
DaylightIsBurning
Они говорят о технологии Molecular Mechanics, которая уже довольно давно применяется в промышленности, в т.ч. фармацевтической.
Это замечание было бы уместно лет 20 лет назад, ну может быть 10, но не сегодня. Сегодня почти ниодно серьезное исследование без компьютерного моделирования не обходится.Desavian
кто спорит? я говорю о том, что результаты компьютерного моделирования нигде не были главными, а пока оно используется только в качестве поддержки — говорить о серьезном уменьшении сроков разработки — рано.
DaylightIsBurning
Не рано. Эта «поддержка» в некоторых случаях уже сейчас позволяет значительно (десятки процентов) сократить время и ресурсы затрачиваемые на экспериментальные испытания. Вместо 100 экспериментов можно обойтись 30ю, скажем, потому что 70 были отсеяны компьютерным моделированием.
arielf
Вы меня опередили! ;-)