
Многабукафф о том, что квантовая химия думает о принципе работы мюонного катализа: как именно мюон понижает температуру требуемой плазмы. В двух частях (первую часть можно прочитать тут).
Суть второй части проста: мюон тяжелее, чем электрон, поэтому он обеспечивает более прочную химическую связь и большее сближение ядер, за счёт чего понижается требуемая температура плазмы для зажигания термоядерной реакции.
Но те, кто хочет посмотреть на формулки, графики, и узреть концептуальную суть квантовой химии в применении к наипростейшим (квази)молекулам, welcome под кат.

В первой части (см. тут) мы разобрали чем отличается атом водорода от своего тяжёлого мюонного аналога : во втором случае мюон будет привязан сильнее, и сидеть он будет на более близком расстоянии от протона. Заодно мы разобрали некоторые важные вещи, которые нам тут понадобятся (формы орбиталей и атомную систему единиц).
Во второй части (т.е. тут) мы попробуем понять, почему, как и насколько понизится температура плазмы, требуемая для зажигания термоядерной реакции. Реакции, что нас интересуют, имеют вид:
где n,m=1,2,3 соответствуют протону, дейтерию и тритию, соответственно. Естественно, эти ядра имеют положительный заряд, поэтому если попробовать их сблизить, они начнут отталкиваться в соответствии с законом Кулона (см. предыдущую часть), и это и есть тот самый барьер, который мешает началу реакций термоядерного синтеза. Кстати, в случае реакций ядерного распада это отталкивание имеет обратную роль, ведь после разъединения из общего ядра, осколки, отталкиваясь друг от друга, приобретают дополнительную кинетическую энергию, и именно этой энергией греется водичка на атомных электростанциях.
На преодоление этого кулоновского барьера и требуется повышение температуры плазмы (T), которая, как все помнят из школьного курса МКТ, связана со средней скоростью частиц в плазме (v) формулой , где m — масса частиц, а — константа Больцмана.
Но, представим себе, что мы объединили два ядра водорода в некую частицу, где они и так расположены близко, и поэтому остаток барьера для них уже очень маленький. Тогда нам требовалось бы существенно меньше разгонять эти частицы (читаем: нужны меньшие температуры), чтобы объединить их в нечто новое. И именно такую роль должен играть промежуточный ион , аналог иона молекулы водорода .
Рассмотрев отличия этих двух частиц, мы поймём, насколько же мюон эффективен в понижении температуры зажигания термоядерного синтеза.
Метод
Итак, у нас есть наша молекулярная система, состоящая из 2-х ядер водорода с зарядом +e (один заряд электрона по модулю) и одной частицы (электрона или мюона) с зарядом –e. Система эта у нас, пока не сталкивается с другими частицами, является изолированной, и поэтому её энергию можно разложить на составные части:
где первые два слагаемых ( и ) — это кинетическая энергия ядер водорода, третий член () — кинетическая энергия отрицательной частицы (электрона или мюона), четвёртый член — это энергия кулоновского отталкивания водородов друг от друга, а оставшиеся два — это кулоновское притяжение электрона/мюона к каждому из протонов. В общем случае — это задача 3-х тел, только ещё квантовая. Естественно, решить её в лоб — очень сложно. Но, к счастью, ядра как минимум в 1800 раз тяжелее электрона, и в 10 тяжелее мюона, поэтому они будут двигаться явно медленнее, чем мелкие отрицательные частицы. За счёт этого можно сначала решать задачу по очереди: сначала находить энергию движений, не связанных с перемещением ядер, т.е. , а потом уже полную энергию. Выглядит это так.
Очевидно, что корень проблемы у нас в вычислении энергии электрона/мюона в поле ядер . По-сути это и есть химическая связь: некий потенциал, который держит ядра вместе на определённых местах. И именно эта задача поиска энергии химической связи и является основной в квантовой химии.
К сожалению, и мюон и электрон являются квантовыми частицами, поэтому, чтобы найти эту энергию, нам придётся прибегнуть к методам квантовой механики. На самом деле наша задача о движении электрона/мюона в поле двух одинаковых ядер решается в явном виде (см. здесь), но это решение очень сложное и результат не такой понятный, как в случае с водородоподобным атомом. Поэтому мы попробуем разобрать другой, приближённый подход, который применим к любым системам. Это т.н. метод молекулярных орбиталей как линейных комбинаций атомных орбиталей, или МО ЛКАО.
Давайте взглянем поподробнее на уравнение Шрёдингера для движения электрона/мюона в поле ядер водорода:
Это уравнение записано в атомной системе единиц (см. P.S. в предыдущей части), поэтому заряд ядра водорода и электрона/мюона равен, соответственно +1, --1, масса электрона m=1, а для мюона m?207.
И если приглядеться, то можно заметить, что в гамильтониане можно выделить кусок, связанный чисто с движением отрицательной частицы вокруг только одного из ядер, что есть просто гамильтониан атома водорода, причём это можно сделать 2-мя способами:
Вне гамильтониана водородоподобного атома () у нас всегда остаются 2 куска: энергия взаимодействия электрона/мюона с другим ядром () и энергия отталкивания ядер (). Вторая из них вообще никак не влияет на движение электронов — это просто сдвиг энергии на некоторую величину, а вот взаимодействие электрона с другим ядром — это вещь важная.
Мы можем вообразить себе, что наша частица в каждый момент вращается только вокруг одного из ядер, а взаимодействие со вторым — это всего-лишь некая поправочка. В качестве способа вращения вокруг одного из ядер можно предположить, что электрон/мюон находится в основном (1s) состоянии, волновая функция для которого хорошо нам известна из предыдущей части:
где — это боровский радиус для частицы. В случае электрона Бор (что и есть боровский радиус для электрона, равный примерно 0.5 ангстрем), а в случае мюона .
Чтобы как-то аппроксимировать волновую функцию электрона/мюона в поле 2х ядер, мы можем попробовать взять следующее представление:
и тогда задача решения сложного уравнения в частных производных у нас сводится к поиску 2х неизвестных коэффициентов c1 и c2. Вот это и есть та самая молекулярная орбиталь, представленная как сумма с коэффициентами (линейная комбинация по-научному) атомных 1s орбиталей.
Естественно, нам нужно уравнение на эти параметры. И получить его достаточно просто, если подставить эту аппроксимацию в уравнение Шрёдингера :
Собственно, мы хотим, чтобы это соотношение выполнялось везде, поэтому можно как-бы посчитать средние значения этого всего. Домножим по очереди слева это уравнение на и и проинтегрируем по всем координатам. В результате мы получим систему из 2-х линейных уравнений, где надо найти коэффициенты c1, c2 и энергию E:
Любой, изучавший линейную алгебру, узнает обобщённую задачу на собственные вектора-собственные значения. Прежде чем её решить, разберём, чему же равны элементы 2х матриц, имеющихся тута (а заодно введём и их краткое обозначение одной буквой).
Найдём же выражения для энергий нашего водородоподобного иона из уравнения, переписанного как
Чтобы найти энергии надо решить уравнение:
где «det» обозначает детерминант (определитель матрицы, по-русски).
Решениями этого квадратного уравнения относительно E являются:
Первый кусок — это, очевидно, энергия атома, второй — межъядерное отталкивание, тот самый кулоновский барьер, мешающий зажиганию термоядерной реакции, а с последней сложной конструкцией надо разобраться.
Если отбросить межъядерное отталкивание, которое всего лишь является точкой отсчёта энергии электрона/мюона, мы получим, что у нас есть два состояния с энергией
Поскольку обе волновые функции и — положительные, а (потому что отрицательную частицу всегда тянет к положительной), то (энергии одинокого атома), а , т.е. мы получаем стандартную картинку молекулярных орбиталей:
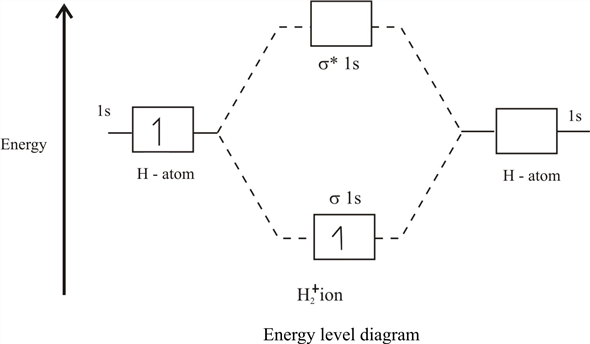
Нижняя орбиталь с энергией называется связывающей, а верхняя (с энергией ) — антисвязывающей, или разрыхляющей. В итоге, если электрон/мюон сидит на нижней молекулярной орбитали, то он получает выгоду от полёта вокруг 2х ядер, чем вокруг одного, и своим движением он понижает общую энергию системы. А это и есть та самая волшебная химическая связь, которая экранирует межъядерное отталкивание, позволяя ядрам находится друг рядом с другом достаточно долгое время.
И вот интегралы химической связи и нужно посчитать, чтобы понять, как близко дозволено находиться ядрам водорода. На самом деле все три искомых интеграла вычисляются аналитически, но это жутко геморройно и сложно (кому интересно, см. главу 9 в книжке «Квантовая химия» Фларри). Поэтому мы пойдём другим путём, более простым, и посчитаем эти интегралы численно при помощи метода Монте-Карло.
Мне видится очень логичным, в тексте про термоядерную энергию воздать почести её дедушке: военному атому, а если конкретнее, Манхэттенскому проекту. Именно из него вырос метод Монте-Карло, и в частности алгоритм Метрополиса, один из авторов которого, Эдвард Теллер, «отец водородной бомбы» (то бишь человек, запустивший термоядерный синтез на атолле Эниветок).
В общем, разберём суть метода. Он предназначен для задач статистической механики. Основным распределением в ней является распределение Больцмана: вероятность обнаружить систему в некотором состоянии равна , . И наблюдаемое значение некоторого параметра A для системы в термодинамическом равновесии равно интегралу
где q — это координаты, параметризующие состояние системы (например, координаты/импульсы частиц), а Z — это нормировочный множитель, называемый статсуммой:
Если в системе ну оооочень много частиц, то посчитать ни один из интегралов в лоб совершенно нереально. Наивный метод Монте-Карло, в котором мы просто выберем кучу случайных значений координат q тоже не даст ничего дельного, если реально возможных состояний системы, для которых вероятность заметно ненулевая, очень мало. И именно для таких случаев нужна выборка по значимости, в которой мы позволим алгоритму сэмплировать только достаточно вероятные места в пространстве состояний.
Выглядит алгоритм Метрополиса следующим образом.
Вот так и работает алгоритм Метрополиса.
А теперь надо бы его адаптировать к вычислению 3-х интересующих нас интегралов. Посмотрим на них поподробнее.
Видно, что, если посчитать 1s-функцию одного из атомов за вероятность p,
то всё остальное под знаком интеграла (вторая волновая функция и в 2-х из 3-х случаев потенциал притяжения электрона/мюона к ядру) будет являться функцией, среднее значение которой вычисляется. Единственное, что придётся делать в отличие от обычного расчёта по методу Метрополиса, так это выправлять нормировку интегралов. Дело в том, что стандартная нормировка у нас будет на
А нам нужна нормировка на , где
Значит, что каждый интеграл, посчитанный по Метрополису, надо будет домножать на множитель
Это уже можно организовать в виде некоего скрипта, например, на Python-е (пример кода есть ниже).
Используя такие вычисления, мы наконец сможем сравнить потенциальные энергии в ионе водорода и его мюонном аналоге.
Итак, вооружившись скриптом, мы можем посчитать поверхности потенциальной энергии сближения ядер водорода, связанных электроном и мюоном. В качестве точки отсчёта энергии возьмём бесконечно разведённые друг от друга атомы (т.е. величину , которой равен потенциал при расстоянии между ядрами ).
В случае электрона потенциал около минимума выглядит так:

Минимум возникает на расстоянии примерно 2 Бора (т.е. примерно сумма 2х атомных радиусов), а энергия диссоциации молекулы на фрагменты приблизительно составляет 0.06 Хартри, что соответствует нагреву до примерно 20000 градусов Кельвина (или Цельсия, тут не важно). Для конвертирования энергий рекомендую пользоваться онлайн ресурсами, типа этого.
Аналогичная ситуация с мюонно связанным ионом водорода:

Поскольку боровский радиус для мюонного водорода меньше (см. предыдущую часть), то и ядра водорода в минимуме потенциальной энергии сидят примерно в 200 раз ближе. Энергия же разрыва этой молекулы составляет уже больше 10 Хартри, что соответствует температуре более трёх лямов градусов ().
Для зажигания же реакции обычно требуют температуру порядка 108 K, что составляет около 320 Хартри. Посмотрим, при каких расстояниях достигается подобная энергия в случае обычного иона диводорода и в случае его мюонной версии:
В случае первого это соответствует расстоянию около 0.0058 Бор (вертикальная линия).
Аналогичное же расстояние в мюонном водороде достигается при энергии около 190 Ха, т.е. примерно в полтора раза меньше. И это и есть простейшая оценка температуры мюонного катализа.
Но на самом деле всё будет ещё круче. Дело в том, что если образовать стабильную частицу , то эти ядра, пока жив мюон, будут колебаться друг относительно друга. И тут может произойти туннелирование из состояния «два атома водорода» в состояние «более тяжёлое ядро», а вероятность туннелирования зависит от необходимой длины туннелирования d примерно как , так что сближая два ядра мюоном мы ооооофигенно сильно увеличиваем вероятность туннельного протекания этой реакции. К сожалению, оценки этого эффекта требуют уже не квантовой химии, а ядрёной ядерной физики, поэтому эта часть рассмотрения выходит за рамки этого поста. Так что на этом мы и остановимся.
На самом деле образовать эти частицы не так уж и просто в условиях плазмы. Дело в том, что если мы сталкиваем две частицы, то их общая энергия заведомо превышает энергию диссоциации (или ионизации, в случае ядра + электрон/мюон), поэтому при столкновении они не образуют стабильную частицу (атом, ион, молекулу), а пролетают мимо друг друга. Чтобы приклеиться друг к другу, им нужно скинуть куда-то излишек энергии, и для этого нужен третий лишний, кто возьмёт на себя эту энергию. Это может фотон, или какая-то левая частица, летающая поблизости, но главное условия должны способствовать этому уносу избытка энергии.
Если есть какие-то замечания/уточнения/вопросы, пишите в комменты или в личку. Всё исправлю, всё отвечу и объясню.
Суть второй части проста: мюон тяжелее, чем электрон, поэтому он обеспечивает более прочную химическую связь и большее сближение ядер, за счёт чего понижается требуемая температура плазмы для зажигания термоядерной реакции.
Но те, кто хочет посмотреть на формулки, графики, и узреть концептуальную суть квантовой химии в применении к наипростейшим (квази)молекулам, welcome под кат.
Введение
В первой части (см. тут) мы разобрали чем отличается атом водорода от своего тяжёлого мюонного аналога : во втором случае мюон будет привязан сильнее, и сидеть он будет на более близком расстоянии от протона. Заодно мы разобрали некоторые важные вещи, которые нам тут понадобятся (формы орбиталей и атомную систему единиц).
Во второй части (т.е. тут) мы попробуем понять, почему, как и насколько понизится температура плазмы, требуемая для зажигания термоядерной реакции. Реакции, что нас интересуют, имеют вид:
где n,m=1,2,3 соответствуют протону, дейтерию и тритию, соответственно. Естественно, эти ядра имеют положительный заряд, поэтому если попробовать их сблизить, они начнут отталкиваться в соответствии с законом Кулона (см. предыдущую часть), и это и есть тот самый барьер, который мешает началу реакций термоядерного синтеза. Кстати, в случае реакций ядерного распада это отталкивание имеет обратную роль, ведь после разъединения из общего ядра, осколки, отталкиваясь друг от друга, приобретают дополнительную кинетическую энергию, и именно этой энергией греется водичка на атомных электростанциях.
На преодоление этого кулоновского барьера и требуется повышение температуры плазмы (T), которая, как все помнят из школьного курса МКТ, связана со средней скоростью частиц в плазме (v) формулой , где m — масса частиц, а — константа Больцмана.
Но, представим себе, что мы объединили два ядра водорода в некую частицу, где они и так расположены близко, и поэтому остаток барьера для них уже очень маленький. Тогда нам требовалось бы существенно меньше разгонять эти частицы (читаем: нужны меньшие температуры), чтобы объединить их в нечто новое. И именно такую роль должен играть промежуточный ион , аналог иона молекулы водорода .
Рассмотрев отличия этих двух частиц, мы поймём, насколько же мюон эффективен в понижении температуры зажигания термоядерного синтеза.
Метод МОЛОКО МО ЛКАО
Итак, у нас есть наша молекулярная система, состоящая из 2-х ядер водорода с зарядом +e (один заряд электрона по модулю) и одной частицы (электрона или мюона) с зарядом –e. Система эта у нас, пока не сталкивается с другими частицами, является изолированной, и поэтому её энергию можно разложить на составные части:
где первые два слагаемых ( и ) — это кинетическая энергия ядер водорода, третий член () — кинетическая энергия отрицательной частицы (электрона или мюона), четвёртый член — это энергия кулоновского отталкивания водородов друг от друга, а оставшиеся два — это кулоновское притяжение электрона/мюона к каждому из протонов. В общем случае — это задача 3-х тел, только ещё квантовая. Естественно, решить её в лоб — очень сложно. Но, к счастью, ядра как минимум в 1800 раз тяжелее электрона, и в 10 тяжелее мюона, поэтому они будут двигаться явно медленнее, чем мелкие отрицательные частицы. За счёт этого можно сначала решать задачу по очереди: сначала находить энергию движений, не связанных с перемещением ядер, т.е. , а потом уже полную энергию. Выглядит это так.
- Выбирается расположение ядер водорода друг относительно друга, и это задаёт кулоновские взаимодействия между ними и с электроном/мюоном. Кулоновский потенциал зависит только от зарядов частиц и расстояния между ними, поэтому для всех изотопов водорода эта величина будет одинакова. Дальше решается задача о движении электрона/мюона в поле этих ядер. Это задача одного тела.
- Эти энергии вычисляются для всех возможных расположений ядер друг относительно друга, и это будет эффективной потенциальной энергией движения ядер. В нашем случае нам нужно посчитать энергии на разных расстояниях друг относительно друга, поэтому потенциал для пары ядер всегда одномерный. Ну и дальше нам нужно только решить задачу двух тел о движении двух изотопов водорода друг относительно друга.
Очевидно, что корень проблемы у нас в вычислении энергии электрона/мюона в поле ядер . По-сути это и есть химическая связь: некий потенциал, который держит ядра вместе на определённых местах. И именно эта задача поиска энергии химической связи и является основной в квантовой химии.
К сожалению, и мюон и электрон являются квантовыми частицами, поэтому, чтобы найти эту энергию, нам придётся прибегнуть к методам квантовой механики. На самом деле наша задача о движении электрона/мюона в поле двух одинаковых ядер решается в явном виде (см. здесь), но это решение очень сложное и результат не такой понятный, как в случае с водородоподобным атомом. Поэтому мы попробуем разобрать другой, приближённый подход, который применим к любым системам. Это т.н. метод молекулярных орбиталей как линейных комбинаций атомных орбиталей, или МО ЛКАО.
Давайте взглянем поподробнее на уравнение Шрёдингера для движения электрона/мюона в поле ядер водорода:
Это уравнение записано в атомной системе единиц (см. P.S. в предыдущей части), поэтому заряд ядра водорода и электрона/мюона равен, соответственно +1, --1, масса электрона m=1, а для мюона m?207.
И если приглядеться, то можно заметить, что в гамильтониане можно выделить кусок, связанный чисто с движением отрицательной частицы вокруг только одного из ядер, что есть просто гамильтониан атома водорода, причём это можно сделать 2-мя способами:
Вне гамильтониана водородоподобного атома () у нас всегда остаются 2 куска: энергия взаимодействия электрона/мюона с другим ядром () и энергия отталкивания ядер (). Вторая из них вообще никак не влияет на движение электронов — это просто сдвиг энергии на некоторую величину, а вот взаимодействие электрона с другим ядром — это вещь важная.
Мы можем вообразить себе, что наша частица в каждый момент вращается только вокруг одного из ядер, а взаимодействие со вторым — это всего-лишь некая поправочка. В качестве способа вращения вокруг одного из ядер можно предположить, что электрон/мюон находится в основном (1s) состоянии, волновая функция для которого хорошо нам известна из предыдущей части:
где — это боровский радиус для частицы. В случае электрона Бор (что и есть боровский радиус для электрона, равный примерно 0.5 ангстрем), а в случае мюона .
Чтобы как-то аппроксимировать волновую функцию электрона/мюона в поле 2х ядер, мы можем попробовать взять следующее представление:
и тогда задача решения сложного уравнения в частных производных у нас сводится к поиску 2х неизвестных коэффициентов c1 и c2. Вот это и есть та самая молекулярная орбиталь, представленная как сумма с коэффициентами (линейная комбинация по-научному) атомных 1s орбиталей.
Естественно, нам нужно уравнение на эти параметры. И получить его достаточно просто, если подставить эту аппроксимацию в уравнение Шрёдингера :
Собственно, мы хотим, чтобы это соотношение выполнялось везде, поэтому можно как-бы посчитать средние значения этого всего. Домножим по очереди слева это уравнение на и и проинтегрируем по всем координатам. В результате мы получим систему из 2-х линейных уравнений, где надо найти коэффициенты c1, c2 и энергию E:
Любой, изучавший линейную алгебру, узнает обобщённую задачу на собственные вектора-собственные значения. Прежде чем её решить, разберём, чему же равны элементы 2х матриц, имеющихся тута (а заодно введём и их краткое обозначение одной буквой).
- Начнём с самого простого: — это нормировка волновых функций, а как мы помним, полная вероятность найти электрон/мюон хоть где-то равна 1.
- — это т.н. интеграл перекрывания, показывающий, насколько перекрываются 1s электронные облака у каждого из атомов.
- . Этот интеграл состоит из нескольких частей:
- . Тут аналогично:
т.е. энергия водородоподобного атома и межъядерного отталкивания, масштабированные на интеграл перекрывания (первый и последний члены), и как бы энергия перескока электрона/мюона с одного атома на другой.
Найдём же выражения для энергий нашего водородоподобного иона из уравнения, переписанного как
Чтобы найти энергии надо решить уравнение:
где «det» обозначает детерминант (определитель матрицы, по-русски).
Решениями этого квадратного уравнения относительно E являются:
Первый кусок — это, очевидно, энергия атома, второй — межъядерное отталкивание, тот самый кулоновский барьер, мешающий зажиганию термоядерной реакции, а с последней сложной конструкцией надо разобраться.
Если отбросить межъядерное отталкивание, которое всего лишь является точкой отсчёта энергии электрона/мюона, мы получим, что у нас есть два состояния с энергией
Поскольку обе волновые функции и — положительные, а (потому что отрицательную частицу всегда тянет к положительной), то (энергии одинокого атома), а , т.е. мы получаем стандартную картинку молекулярных орбиталей:
Нижняя орбиталь с энергией называется связывающей, а верхняя (с энергией ) — антисвязывающей, или разрыхляющей. В итоге, если электрон/мюон сидит на нижней молекулярной орбитали, то он получает выгоду от полёта вокруг 2х ядер, чем вокруг одного, и своим движением он понижает общую энергию системы. А это и есть та самая волшебная химическая связь, которая экранирует межъядерное отталкивание, позволяя ядрам находится друг рядом с другом достаточно долгое время.
И вот интегралы химической связи и нужно посчитать, чтобы понять, как близко дозволено находиться ядрам водорода. На самом деле все три искомых интеграла вычисляются аналитически, но это жутко геморройно и сложно (кому интересно, см. главу 9 в книжке «Квантовая химия» Фларри). Поэтому мы пойдём другим путём, более простым, и посчитаем эти интегралы численно при помощи метода Монте-Карло.
Метод Метрополиса
Мне видится очень логичным, в тексте про термоядерную энергию воздать почести её дедушке: военному атому, а если конкретнее, Манхэттенскому проекту. Именно из него вырос метод Монте-Карло, и в частности алгоритм Метрополиса, один из авторов которого, Эдвард Теллер, «отец водородной бомбы» (то бишь человек, запустивший термоядерный синтез на атолле Эниветок).
Про принцип Арнольда
Несмотря на то, что алгоритм носит имя Метрополиса, последнего (и типа главного) автора оригинальной статьи «Equation of State Calculations by Fast Computing Machines» в Journal of Chemical Physics, разные источники утверждают, что автором алгоритма является Энрико Ферми, его переоткрыл Стэн Улам (обоих вообще нет в авторах), или же Маршал Розенблют, стоящий на 3м месте в списке авторов, а Николас Метрополис же является автором термина «Монте-Карло», и человеком, предоставившим компьютерное время. Всё в полном соответствии с принципом Арнольда. :D
В общем, разберём суть метода. Он предназначен для задач статистической механики. Основным распределением в ней является распределение Больцмана: вероятность обнаружить систему в некотором состоянии равна , . И наблюдаемое значение некоторого параметра A для системы в термодинамическом равновесии равно интегралу
где q — это координаты, параметризующие состояние системы (например, координаты/импульсы частиц), а Z — это нормировочный множитель, называемый статсуммой:
Если в системе ну оооочень много частиц, то посчитать ни один из интегралов в лоб совершенно нереально. Наивный метод Монте-Карло, в котором мы просто выберем кучу случайных значений координат q тоже не даст ничего дельного, если реально возможных состояний системы, для которых вероятность заметно ненулевая, очень мало. И именно для таких случаев нужна выборка по значимости, в которой мы позволим алгоритму сэмплировать только достаточно вероятные места в пространстве состояний.
Выглядит алгоритм Метрополиса следующим образом.
- При инициации симуляции мы выбираем некоторое стартовое приближение в пространстве конфигураций и некоторый вектор максимально возможного приращения . В стартовой точке вычисляем энергию системы (читаем — вероятность ).
- Новая конфигурация на n-м шагу получается так.
- Вычисляем энергию пробной конфигурации (т.е. вероятность ).
- А дальше сравниваем между собой старую вероятность с пробной
- если новая конфигурация имеет большую или такую же вероятность (), или, что эквивалентно, энергия новой точки ниже или та же, что и в старой (), то новая точка принимается и система переходит в неё (),
- если же пробная конфигурация выше по энергии (), что эквивалентно , то в этом случае мы генерируем случайное число из равномерного распределения, и сравниваем его с отношением вероятностей, которые являются вероятностью перехода. Если , то мы принимаем новую точку, а если нет (), то отвергаем, и система остаётся в старой конфигурации ()…
- Делая много шагов по алгоритму выше, мы и сэмплируем значимую (т.е. реально важную) часть возможного пространства конфигураций системы. Интересующий же нас интеграл вычисляется по формуле:
Вот так и работает алгоритм Метрополиса.
А теперь надо бы его адаптировать к вычислению 3-х интересующих нас интегралов. Посмотрим на них поподробнее.
- , где — координаты электрона/мюона, — координаты ядер водорода, а — расстояния между положительными и отрицательными частицами,
Видно, что, если посчитать 1s-функцию одного из атомов за вероятность p,
так делать, конечно же не очень хорошо,
потому что плотность вероятности — это модуль квадрата волновой функции , а не сама волновая функция .
то всё остальное под знаком интеграла (вторая волновая функция и в 2-х из 3-х случаев потенциал притяжения электрона/мюона к ядру) будет являться функцией, среднее значение которой вычисляется. Единственное, что придётся делать в отличие от обычного расчёта по методу Метрополиса, так это выправлять нормировку интегралов. Дело в том, что стандартная нормировка у нас будет на
А нам нужна нормировка на , где
Значит, что каждый интеграл, посчитанный по Метрополису, надо будет домножать на множитель
Это уже можно организовать в виде некоего скрипта, например, на Python-е (пример кода есть ниже).
Например, так.
import numpy as np
from math import *
# r = 0...+infty
# phi = 0...2pi
# theta = 0...pi
# function to convert spherical coordinates into Cartesian
def sph2cart(r, phi, theta):
xyz=np.zeros(3)
xyz[0]=r*cos(phi)*sin(theta)
xyz[1]=r*sin(phi)*sin(theta)
xyz[2]=r*cos(theta)
return xyz
# Distance between vectors r1 and r2
def dist(r1, r2):
return sqrt(np.dot(r1-r2, r1-r2))
# re -- Cartesian coordinates of electron
# rn -- Cartesian coordinates of nucleus
# psi_1s returns a value of 1s wavefunction
def psi_1s(re, rn, scale=1.0):
ren=dist(re,rn)
return scale**(3/2)*exp(-scale*ren)/(sqrt(pi))
########################################
############ Settings ##################
########################################
NumPtsPerIntegral=100000 # Number of points per integral... duuuh
#mass=1.0 # mass of the particle (electron)
mass=207.0 # mass of the particle (muon)
AllRab=[]
#AllRab+=[1.0*(0.1)**n for n in range(0,10) ]
AllRab+=[ (1.4+0.25*n)/mass for n in range(0,10)]
print(AllRab)
########################################
########################################
########################################
dumpster=open("res.dat", "w") # output file to store results of the simulation
dx=2.0/mass # maximal increment for the coordinate
rna=np.array([0.0, 0.0, 0.0]) # position of nucleus "a"
renorm=8.0*sqrt(pi/mass**3) # factor to readjust result from incorrect norm of Metropolis weighting to a correct 1s wavefunction norm
# loop for the potential energy calculation at the chosen internuclear distances
for npt,Rab in enumerate(AllRab):
Norm=0.0 # <1s_a | 1s_a > for check
Sab=0.0 # <1s_a | 1s_b >
Vaa=0.0 # <1s_a | |r - R_b|**(-1) | 1s_a >
Vab=0.0 # <1s_a | |r - R_b|**(-1) | 1s_b >
re=np.array([1.0/mass, 0.0, 0.0]) # initial position of the electron
rnb=np.array([Rab, 0.0, 0.0]) # position of nucleus "b"
NumAcc=0.0 # Number of accepted points
for i in range(0,NumPtsPerIntegral): # loop for Metropolis algorithm
newre=re+np.random.uniform(low=-dx, high=dx, size=3) # trial position of electron
pnew=psi_1s(newre, rna, scale=mass) ## trial probability
pold=psi_1s(re, rna, scale=mass) ## previous probability due to dumb and ineffective realization
if pnew/pold >= np.random.random(): ## importance sampling step
re=newre
NumAcc+=1.
Norm+=psi_1s(re, rna, scale=mass)
Sab+=psi_1s(re, rnb, scale=mass)
Vaa+=psi_1s(re, np.zeros(3), scale=mass)/dist(re, rnb)
Vab+=psi_1s(re, rnb, scale=mass)/dist(re, rnb)
Norm*=renorm/NumPtsPerIntegral
Sab*=renorm/NumPtsPerIntegral
Vaa*=renorm/NumPtsPerIntegral
Vab*=renorm/NumPtsPerIntegral
def s_test(x,scale=1.0): # this is an analytical expression for overlap integral S in case of 1s hydrogen wavefunctions
return exp(-x*scale)*(1.+x*scale+(1./3.)*(x*scale)**2)
#E=-0.5*mass**2 + 1./sqrt(np.dot(rnb,rnb)) - (Vaa + Vab)/(1.0 + Sab) # full energy
E= 1./sqrt(np.dot(rnb,rnb)) - (Vaa + Vab)/(1.0 + Sab) # energy adjusted to energy of a single atom as the dissociational limit
dumpster.write(" %10.3e %40.10f %15.10f %15.10f %15.5f %15.5f\n" % (Rab, E, Sab, s_test(Rab,scale=mass), 100.0*NumAcc/NumPtsPerIntegral, Norm ))
dumpster.flush()
dumpster.close()
Используя такие вычисления, мы наконец сможем сравнить потенциальные энергии в ионе водорода и его мюонном аналоге.
vs.
Итак, вооружившись скриптом, мы можем посчитать поверхности потенциальной энергии сближения ядер водорода, связанных электроном и мюоном. В качестве точки отсчёта энергии возьмём бесконечно разведённые друг от друга атомы (т.е. величину , которой равен потенциал при расстоянии между ядрами ).
В случае электрона потенциал около минимума выглядит так:
Минимум возникает на расстоянии примерно 2 Бора (т.е. примерно сумма 2х атомных радиусов), а энергия диссоциации молекулы на фрагменты приблизительно составляет 0.06 Хартри, что соответствует нагреву до примерно 20000 градусов Кельвина (или Цельсия, тут не важно). Для конвертирования энергий рекомендую пользоваться онлайн ресурсами, типа этого.
Аналогичная ситуация с мюонно связанным ионом водорода:
Поскольку боровский радиус для мюонного водорода меньше (см. предыдущую часть), то и ядра водорода в минимуме потенциальной энергии сидят примерно в 200 раз ближе. Энергия же разрыва этой молекулы составляет уже больше 10 Хартри, что соответствует температуре более трёх лямов градусов ().
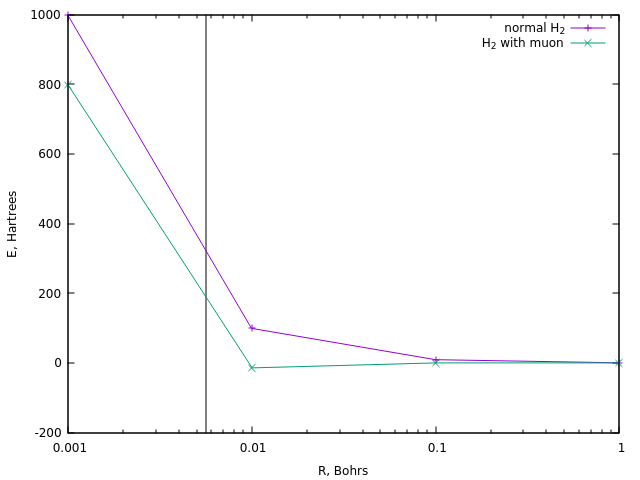
Для зажигания же реакции обычно требуют температуру порядка 108 K, что составляет около 320 Хартри. Посмотрим, при каких расстояниях достигается подобная энергия в случае обычного иона диводорода и в случае его мюонной версии:
В случае первого это соответствует расстоянию около 0.0058 Бор (вертикальная линия).
Аналогичное же расстояние в мюонном водороде достигается при энергии около 190 Ха, т.е. примерно в полтора раза меньше. И это и есть простейшая оценка температуры мюонного катализа.
Но на самом деле всё будет ещё круче. Дело в том, что если образовать стабильную частицу , то эти ядра, пока жив мюон, будут колебаться друг относительно друга. И тут может произойти туннелирование из состояния «два атома водорода» в состояние «более тяжёлое ядро», а вероятность туннелирования зависит от необходимой длины туннелирования d примерно как , так что сближая два ядра мюоном мы ооооофигенно сильно увеличиваем вероятность туннельного протекания этой реакции. К сожалению, оценки этого эффекта требуют уже не квантовой химии, а ядрёной ядерной физики, поэтому эта часть рассмотрения выходит за рамки этого поста. Так что на этом мы и остановимся.
P.S. Почему не всё так просто?
На самом деле образовать эти частицы не так уж и просто в условиях плазмы. Дело в том, что если мы сталкиваем две частицы, то их общая энергия заведомо превышает энергию диссоциации (или ионизации, в случае ядра + электрон/мюон), поэтому при столкновении они не образуют стабильную частицу (атом, ион, молекулу), а пролетают мимо друг друга. Чтобы приклеиться друг к другу, им нужно скинуть куда-то излишек энергии, и для этого нужен третий лишний, кто возьмёт на себя эту энергию. Это может фотон, или какая-то левая частица, летающая поблизости, но главное условия должны способствовать этому уносу избытка энергии.
P.P.S.
Если есть какие-то замечания/уточнения/вопросы, пишите в комменты или в личку. Всё исправлю, всё отвечу и объясню.
Eugeny1987
flx0
Мне вот тут одна идея пришла на счет мюонного катализа, не знаю насколько адекватная.
Нельзя ли с его помощью получать металлический водород? Берется кусок твердого замороженного водорода (вероятно в виде довольно тонкой пленки), рядом ставится анод, чтобы забирать с него электроны, с противоположного конца обстреливается мюонами. Они вытесняют электроны, атомы в куске теряют в размере и становятся ближе, потом мюоны распадаются на те же электроны и нейтрино. А атомы уже ближе, и без всякого давления сливаются в металлическую решетку.
Или это совсем не так работает?
madschumacher Автор
Белые хлопушки при температуре ?259,2 °C (14,16 К). Сомневаюсь, что можно их в тонкую плёнку превратить.
По-моему ощущению, мюоны просто будут ускоряться анодом и проходить через вещество, попутно дифрагируя на нём.
В этих условиях это неравновесное состояние, при малейшей флуктуации оно в соответствии с фазовой диаграммой:
вернётся к стабильной форме.
sinc
для того чтобы ионизировать водород нужно около 3000 К. почему с расчетом так далеко?
madschumacher Автор