
Мы уже несколько раз упоминали серию мероприятий Data & Science, где специалисты по анализу данных и учёные рассказывают друг другу о своих задачах и ищут способы для взаимодействия. Одна из встреч была посвящена биоинформатике. Это отличный пример отрасли, где есть масса ещё не решённых задач для разработчиков.

Под катом вы найдёте расшифровку лекции Игната Колесниченко — выпускника мехмата МГУ и Школы анализа данных. Сейчас Игнат работает ведущим разработчиком службы технологий распределённых вычислений Яндекса.
Кто здесь программист? Примерно половина аудитории. Мой доклад — для вас.
Цель доклада следующая: я в точности расскажу, как берутся данные и как они устроены. Рассказ будет чуть более глубоким, чем у Андрея. Для этого придется немного напомнить про биологию. Я, по крайней мере, рассчитываю, что программисты не знают биологию.
А дальше попробую рассказать, какие алгоритмически сложные задачи решаются в данной области, чтобы заинтересовать вас, чтобы вы поняли, что это здорово и надо идти в биоинформатику, помогать людям решать сложные задачи. Очевидно, сами биологи с алгоритмическими задачами не справятся.
Я окончил мехмат. Я математик, работаю в Яндексе. Я программист, занимаюсь системой YT, мы делаем внутреннюю инфраструктуру в Яндексе, у нас кластера на тысячи машин. Все данные, которые собирает Яндекс, хранятся в этих кластерах, всё обрабатывается, петабайты данных, в этом я немного понимаю. А еще я сооснователь компании «Эбином». Там я занимался такой же технической задачей. Моя цель была — разобраться, как работают эти алгоритмы, программы, и научить их делать это наиболее автоматизированно, чтобы человек мог залить данные на сайт, ткнуть кнопку, и у него бы развернулся кластер, на нём всё быстренько бы посчиталось, свернулось, и данные бы показались человеку.
Пока я решал эту техническую задачу, мне приходилось разбираться, о каких данных идет речь и как с ними работать. Сегодня я расскажу часть того, что узнал.
На Фейсбуке в анонсе к этому мероприятию было выложено видео Гельфанда. Кто его смотрел? Один человек. Гельфанд смотрит на вас неодобряюще.
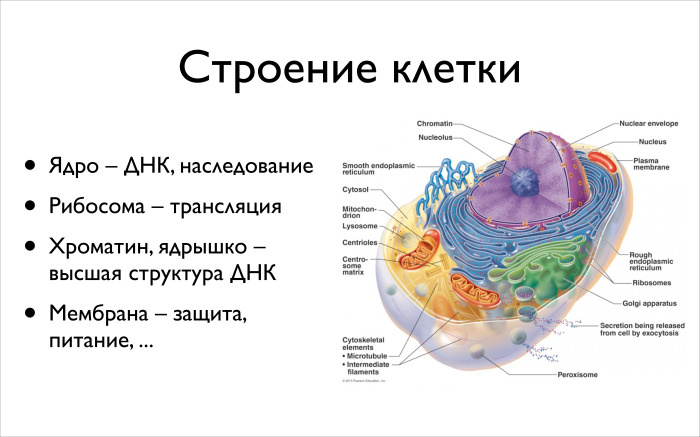
Мы будем работать с ДНК, но хочется немного контекста вокруг ДНК, так что мы начнем с клетки. Понятно, что биоинформатика как наука о молекулярной биологии интересуется всеми процессами в клетке, но мы сегодня будем больше разговаривать про техническую сторону, про данные и нас будут интересовать, условно, лишь ядро и близлежащие компоненты.
В ядре содержится ДНК. В нем есть ядрышко. Оно вместе с гистонами, которые показывал Андрей, в некотором смысле отвечает за регуляцию и эпигенетику. Есть вторая важная часть — рибосома, складочки вокруг ядра, так называемый эндоплазматический ретикулум. В нем есть специальные комплексы молекул, называемые рибосомами. Оно будут из ваших кусочков ДНК, которые вы сначала из ДНК вычитаете, делать белки.
Третья часть — хроматин: всё вещество, в котором всё ДНК плавает, находится. Оно упаковано достаточно плотно, но между ним всё равно ещё есть много разных регулирующих молекул. Ну и есть мембрана — защита, питание, то, как клетка общается.
Еще в клетке есть многое другое, другие функции. Про них мы говорить не будем, можете почитать в учебниках.
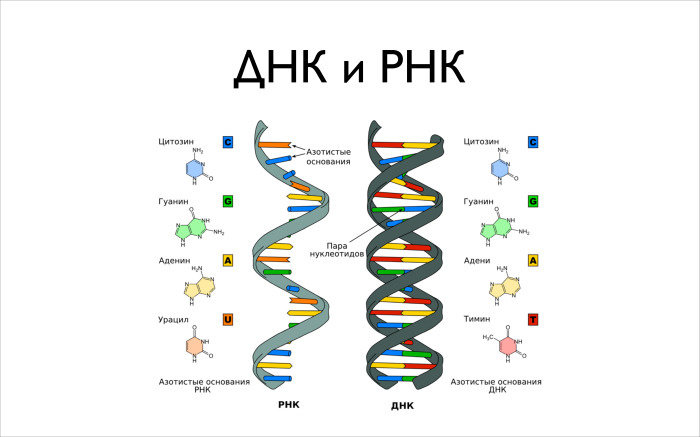
Теперь про информацию, ДНК и РНК. Есть две основных молекулы: дезоксирибонуклеиновая кислота и рибонуклеиновая кислота. Это молекулы одного типа. Когда-то Уотсоном и Маккриком была открыта структура этой молекулы. Интересная история, можете почитать автобиографию, он очень красиво про всё пишет. Она представляет собой двойную спираль, это популярная картинка, популярное buzzword.
Состоит она из четырех более крупных молекул, комплексов из десятков атомов. Это гуанин, цитозин, аденин и тимин. Причем в РНК вместо тимина будет урацил. Когда вы из ДНК будете делать РНК, вместо буквы «Т» вы получите букву «У». Нам важно знать, что в нашем ДНК содержатся эти четыре буквы. С ними мы и будем работать, по большому счету.
У ДНК есть высшая структура. Это не просто спиралька, которая как-то лежит, абы как скручена. Она закручивается вокруг специальных комплексов — гистонов. Потом гистоны закручиваются в некоторые волокна, из которых уже строится буква Х. Перед вами красивая картинка, которую вы видели в учебниках биологии. Обычно про гистоны не говорят, 15 лет назад не говорили, а показывают букву Х. Она устроена таким образом, ДНК там хитрым образом закручено.
Гистоны отвечают за регуляцию. Когда вы захотите прочитать какую-то РНК с ДНК, у вас может не получиться, потому что оно так закрутилось, так спрятано в этой ДНК, что оттуда не читается. А может, что-то поменяется и оно начнет читаться.
И есть разные белки, которые умеют ко всему этому привязываться, умеют это разворачивать, вытаскивать, чтобы вы прочитали. Вторичная структура очень важна, чтобы понимать, какие участки ДНК, из которых вы потом можете делать РНК белки, вам доступны для чтения.
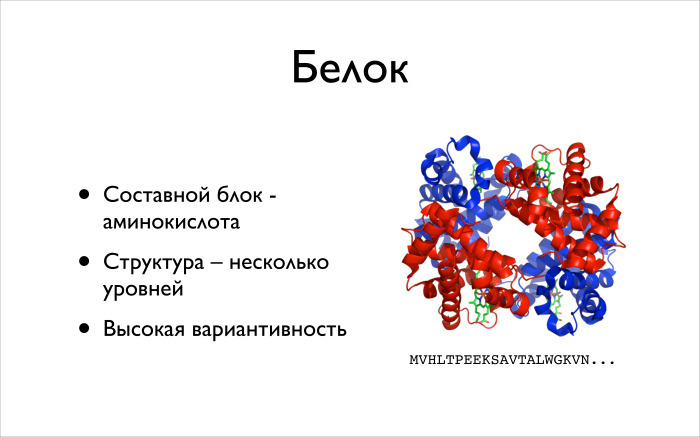
Что такое белок? Основная жизненная сила клетки, многообразие всего живого есть во многом благодаря белкам. Дело в том, что у белка очень высокая вариативность. Речь идет об огромном классе очень разнообразных молекул, которые участвуют в огромной куче молекул. Они имеют очень разные структуры, что и позволяет всей этой сложной химии происходить внутри клетки.
Что нужно знать про белки? Составной блок — аминокислота. Аминокислот в нашем организме, в нашем мире, 20 штук. Говорят, их немножко больше, но в нашем организме их 20. У них тоже есть определенные латинские буквы. Справа нарисован белок схематически, выписано его начало, указано, какие буквы присутствуют. Я не биолог, не помню, что такое V или M, а биологи сразу могут сказать, что буквы означают.
Структура белка имеет несколько уровней, можно на нее смотреть просто как на последовательность, и когда вы биоинформатик, вы часто именно так и смотрите. Хотя вот Артур интересуется более сложными вторичными и третичными структурами.
Мы будем смотреть на это как на последовательность букв. Вторичная и третичная структуры. Есть такие спиральки, альфа-спирали, они на картинке хорошо видны. Есть еще бета-листы, она умеет в достаточно плоские штуки выстраиваться, и есть специальные штуки, которые собирают эти альфа-спирали и бета-листы.
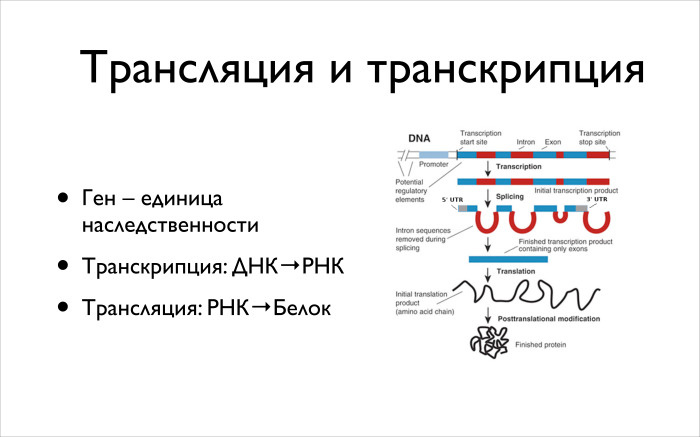
Дальше то, о чем говорил Андрей, — центральная догма молекулярной биологии. Она состоит в том, что есть трансляция и транскрипция, а есть еще много разных процессов обратных, перпендикулярных, которые тоже задействуют РНК, ДНК и белки в разных направлениях. Но мы посмотрим сегодня только на трансляцию и транскрипцию. Справа есть достаточно полная картинка, описывающая эту вещь в эукариотических клетках, в частности в людях.
Есть у вас ген. В учебниках биологии пишут, что это единица наследственности. Для нас же, когда мы смотрим на ДНК, ген — некоторый не такой простой участок. Не то что он транслируется в РН, а дальше из РНК делается белок. Происходит всё немного не так. Во-первых, в указанном участке есть регионы, интроны, которые вырезаются. А есть экзоны, которые потом и будут кодироваться в ваш белок. Если вы посмотрите на ДНК, на 3 млрд букв, то — вот популярная фраза — важных областей там всего 2%. Только 2% отвечают за кодирование ваших генов. И речь идет именно про экзоны.
Если вы возьмете еще интроны, у вас получится не 2%, а 20%. А оставшиеся 80% — это межгенные области, участвующие, по всей видимости, в какой-то степени в регуляции. Но некоторые действительно низачем не нужны, просто так исторически сложилось.
Происходит следующее: у вас есть некий промоутер перед геном, на этот промоутер садится специальная молекула, белок, который будет считывать и из вашего ДНК делать РНК.
Дальше из РНК преобразование идет другой молекулой, которая из него вырезает ненужные интроны. Вы получаете уже готовую матричную РНК, из которой делаете ваш белок трансляцией. При этом отрезаются 3’ и 5’ — некодируемые концы. Когда из РНК вы делаете белок, на самом деле у вас есть триплет, некоторая последовательность, с которой вы можете начать. Всё, что до неё, просто пропускается. Клетка садится, она это пропускает, и начинает всё делать со стартового кодона.
Я употребил слово кодон. А что такое кодоны? как происходит кодирование? Логика простая. В ДНК есть четыре символа: A, C, T, G. В белке 20 символов, 20 аминокислот. Надо как-то из четырех сделать 20. Понятно, что напрямую не получится, надо их как-то закодировать. С точки зрения ученых, которые занимаются кодированием, устройство очень простое, глупое кодирование, мы бы такое легко придумали. Но с точки зрения биологии выполнить такое было непросто.
У вас читается по три буквы ДНК, и каждый триплет букв кодирует определенную аминокислоту. Триплетов букв 64, и 20 аминокислот можно закодировать 64 вариантами. Кодирование получится избыточным. Есть разные триплеты, которые кодируют одно и то же.
Это всё, что нужно знать на сегодня про трансляцию и транскрипцию.
Андрей много говорил про метагеномику, про разные омики. Ученые последние 50–100 лет очень активно изучают, что происходит в клетке, какие там процессы, какая там химия, а не только биология. И они достаточно сильно преуспели в этом вопросе. Может, у них нет совсем полной картины и понимания, но очень многие вещи они знают.
Есть большие карты преобразования разных веществ в друг друга. В частности, большинство элементов, белки, которые описывают, что вообще в клетке происходит.

Вот маленький кусочек, а есть большая картинка, ученые любят ее рисовать. Перед нами примерная упрощенная схема всего, что делается в нашей клетке.
Важно понимать, что все молекулы более-менее всегда в нашей клетке присутствуют — вопрос в концентрациях. Все время ваше ДНК где-нибудь разворачивается, что-нибудь из нее читается, и то, что читается, во многом зависит от того, какая концентрация у разных белков. И вся жизнь клетки — регуляция. У вас чего-нибудь стало больше, это что-нибудь пошло делать больше другого процесса, он начал больше что-то транслировать, появился новый белок, он поучаствовал в каком-нибудь процессе, сказал, что хватит меня производить, породил новый белок, который стал мешать его производству. Такие зацикленные с обратной связью процессы в огромном количестве присутствуют в клетке и здесь в целом представлены.
Откуда берутся данные — эти A, C, T, G? Андрей говорил, что есть секвенаторы, которые вам их прочитают. Давайте поймем подробнее, как эти приборы устроены и что они прочитают.
Есть классический метод Сенгера, который придумали в конце 70-х, и которых позволил в 1989 году начать проект «Геном человека». К 2001 году проект завершили, смогли собрать последовательность некоего «эталонного» человека, состоящую из 3 млрд букв. Мы разобрали все 23 хромосомы и поняли, какие буквы — A, C, T, G — в каждой хромосоме находятся.
Метод Сенгера хороший и надежный, ученые ему доверяют, но он очень медленный. В 1977 году это был страшная жуть, вы работали с некоторыми радиоактивными растворами и веществами в перчатках, находясь за свинцовой штучкой, что-то куда-то заливали в пробирке. Через три часа после этих упражнений, если вы случайно не туда не вылили что-то, вы получали картинку, из которой могли узнать 200 нуклеотидов вашего ДНК.
Понятно, что процесс быстро автоматизировали, и в ходе автоматизации технологии немного поменялись. К середине 2000-х появилась масса новых технологий, которые позволяют делать секвенирование быстро и дешево и не тратить на каждые 400 символов три часа работы дорогостоящего ученого.
Последняя технология, о которой говорил Андрей, — мининанопоры. Мне кажется, в научном сообществе пока есть некоторое сомнение насчет того, станет ли эта технология мейнстримом или нет. Но она точно открывает для нас новые границы.
Все предыдущие технологии считают достаточно точно, в них ошибок мало, речь идет про проценты и доли процентов. В этой технологии ошибок существенно больше, 10-20%, но вы можете читать большие иды. Кажется, в больших приложениях это действительно необходимо.
Сумеют ли это добить до технологии, в которой тоже будет мало ошибок, — неизвестно. Но если смогут, будет очень круто.
Общая картина про секвенирование, про то, как это происходит. У вас берут пробу, анализ крови, анализ слюны, или залезают специальным пинцетом, достают кусочек костного мозга: очень болезненная страшная процедура. Дальше по пробе делается некоторая химия, которая все лишнее удаляет и оставляет, условно, только ваше ДНК, которое в пробе присутствовало.
Дальше вы делаете фрагментацию ДНК, получаете маленькие фрагменты, которые вы дальше размножаете, чтобы их было побольше. Это нужно во многих технологиях. Затем кормите секвенатор, он всё прочитывает некоторым способом.
На выходе — всегда одинаковый fastq-файл. Все приборы работают по-разному, у них разные длины, разные типы ошибок, и если вы будете работать с этими данными, то важно понимать, какие здесь есть типы ошибок, как с такими данными правильно работать. Если вы будете применять один и тот же метод ко всему, вы будете получать грязные данные, на которые нельзя положиться.
Кратко про метод Сенгера. Идея очень красивая. У вас есть ДНК-последовательность, вы ее расплодили, она плавает у вас в пробирке. Дальше вы хотите понять, из чего она состоит. Туда скармливаются специальные белки. Может, там даже будет плавать вся ДНК, но будет плавать и молекула, которая умеет ее разбирать.
У ученых очень простой способ. Как они такие вещи с ДНК научились делать? Они посмотрели, как то же самое делает природа в наших клетках, поняли, какие белки что делают, достали их, и теперь сами могут реплицировать ДНК. Просто берут белки, которые занимаются репликацией, и кидают их в пробирку. Там все само происходит.
Они кидают эти белки, и еще кидают наши ACTG. Среди них есть специальные помеченные, которые, когда она приклеилась при чтении к нашей ДНК, дальше не позволяет клеить еще. Накидав все это дело, взболтнув и дав немного постоять, мы получаем кусочки недочитанной исходной ДНК, которой мы интересуемся. У нас случайным образом что-нибудь приклеивается, и надежда в том, что для каждой длины у нас что-нибудь наклеится, вот в этот момент останавливающая ТТР наклеится и всё нам остановит.
Ещё специальные помеченные ACTG, флуоресцентные. Если на них посветить специальным образом, они будут зелеными, синими, красными или желтыми. Затем все это бросают в гель, там они по массе распределяются, их подсвечивают и получают картинку, как справа. Дальше по ней можно посчитать сверху вниз: более короткие выше, более тяжелые ниже.

В итоге мы получаем fastq-файл. Как он выглядит, нарисовано справа. Это такой текстовый формат, что уже должно программистов вводить в недоумение — зачем хранить в текстовом виде, когда можно сжимать? Вот так сложилось. По факту хранят в таком файлике, просто жмут его каким-нибудь KZIP. В файлике записаны данные в четырех строчках: сначала идет ID вашего одного рида, прочтения. Дальше буквы, которые там прочитались. Потом некоторая техническая строка, и в четвертой строке идет качество. Все приборы умеют выдавать некоторые показатели того, насколько они уверены в прочитанном. Дальше, когда вы будете работать с данными, это надо учитывать.
Суть работы большинства алгоритмов в том, что мы читаем много-много раз с большим покрытием случайные кусочки. Дальше по ним хотим понять, как выглядела исходная ДНК. Понятно, что когда люди начинали проект «Геном человека», никаких других собранных геномов у них толком не было. Бактерии тогда уже научились собирать. И это была страшная задача. У вас есть геном человека, в нем 3 млрд символов. Вы получили кучу кусочков длины 300-400, и по ним надо теперь понять, какая же была последовательность, из которой такие кусочки могли получиться. Выглядит страшно.
Поговорим про алгоритмы. Какие задачи решаются с помощью NGS и как они решаются с алгоритмической точки зрения?

Первая задача — сборка генома. Про нее можно прочитать полугодовой курс лекций, там очень много материалов. Я набросаю только общие идеи, чтобы вы поняли, какие алгоритмы здесь применяются, почему это сложно и интересно.
В чем состоит задача сборки генома? Даны риды в формате fastq, их миллионы или десятки миллионов. Например, вы собираете одну хромосому. А надо вам получить геном или хромосому, которую вы хотите собрать.
Теперь, с программистской точки зрения, как формулируется это задача? У вас есть короткие строки из символов ACTG, на них есть некоторые показатели их качества, веса. И когда вы их собираете, есть еще ограничения на ответ. Когда биологи собирали геном человека, они знали, что на 11 хромосоме есть такие-то гены: мы их в результате долгой работы исследовали и другими способами выяснили, что они там есть. И это хочется учитывать. Было бы глупо собрать что-нибудь, а потом выяснить — того, в чем мы уверены, почему-то нет, поскольку алгоритм так решил.
Надо получить некую суперстроку над всеми короткими строками. Причем мы хотим, чтобы она была как можно меньше. Иначе мы можем просто склеить все наши фрагменты и получить что-то, что никому не годится. Поскольку все строки на входе были получены из нашей хромосомы, значит, надо найти эту хромосому. Если мы делали это с достаточно большим покрытием — условно, с каждым сдвигом есть какая-то строчка — и мы соберем самую короткую, это и будет наш геном. Сказанное не всегда правда, есть разные причины, почему можно что-то потерять или упустить в такой формулировке. Но это достаточное приближение, которое всех устраивает.

Упрощенный пример задачи. Можно собрать суперстроку, которая будет короче, чем их длина 16, и она может выглядеть вот так. Мы взяли последнюю строку ССТА, к ней приклеили предпоследнюю, буквы ТА у них перекрылись. Мы, таким образом, два символа сэкономили. Потом к АС приклеили первую строчку. Вторая строчка TGAC оказалась неудачной, ее пришлось просто так приклеить. Понятно, что пример искусственный. По факту таких приклеиваний в настоящем мире у вас не будет.
Есть плохие новости для вас. Те, кто работает программистом и разбирается в алгоритмах, могут открыть стандартный учебник Гэри Джонсона про сложность алгоритмов и выяснить, что задача о суперстроке является NP-трудной. Кто не знает, на практике это примерно означает, что никакого полиномиального алгоритма решения задачи ожидать не приходится: все известные алгоритмы имеют экспоненциальное время работы. А это оказывается полный ужас: у вас десятки миллионов строк, и как вы за экспоненциальное время будете что-то делать? 2 в степени 10 млн — такого за время жизни Вселенной не дождешься.
Но есть и хорошая новость. у нас не худший вход для задачи, какой-то из вполне реальных. И для него есть надежда решить задачу достаточно хорошо.
Общий подход к решению в следующем: по факту нам не удастся разом собрать всю хромосому непрерывно. Тому есть разные причины. Кажется, проще их не объяснять, а смириться, что так и будет.
Когда мы начнем собирать, мы соберем достаточно большие кусочки из сотен тысяч символов, может даже из миллионов. Дальше нам придется их склеить во всю хромосому размером 100 млн. Эти непрерывные кусочки, которые получится собрать, называются контигами. Мы потом по разным вторичным данным глобально пытаемся понять, как контиги друг с другом соотносятся, и собираем их scaffold’ом. В нашем случае речь идет о хромосоме, если мы собираем хромосому человека.
Есть два алгоритмических метода. Это был конец 80-х и начало 90-х, золотой век людей, которые занимались строковыми алгоритмами. Они всю свою теорию и науку придумали в конце 60-х и начале 70-х, но никому она было не нужна. Я даже был на докладе человека, который придумал суффиксное дерево. Он был только ученым-аспирантом, в аспирантуре придумал суффиксное дерево, написал статью и потом ушел в бизнес. И рассказывал, как в конце 80-х неожиданно ему стали писать письма биологи и спрашивать, как в вашей статье это и это работает. Он был очень удивлен, очень рад за них. Очень вдохновил этот случай.
Зачем все понадобилось? Один из алгоритмов называется Overlap-Layout-Consensus. Мы хотим построить на наших кусочках overlay-граф, дальше мы хотим этот граф упростить, выкинув из него все ненужное, и дальше мы в целом поймем, как будут устроены наши контиги.
Overlay-граф — следующая вещь. Есть у нас два наших рида, мы хотим для каждой — что уже звучит немного безумно с учетом наших данных — пары наших ридов понять, насколько сильно они перекрываются.
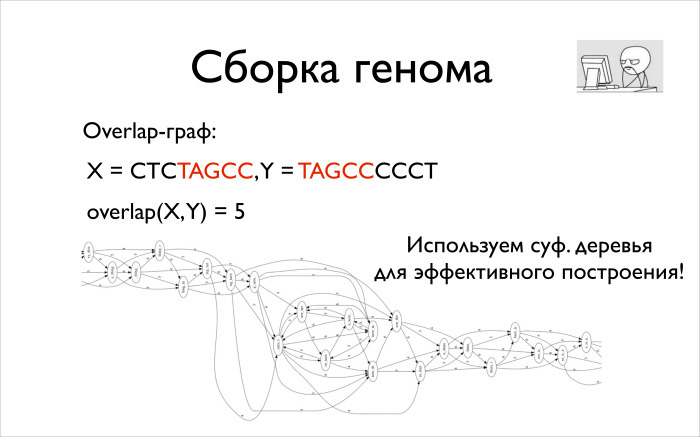
Есть такие риды. Они перекрываются на 5, и красным видно, где они перекрываются.
Если мы построим граф этих перекрытий, дальше можно будет с ним работать, пытаться прочитать по этому графу наш геном. Вопрос, как это делать, — отдельный. Очевидно, сразу встает задача для каждой пары ридов построить overlap. У нас ридов ну пусть даже миллион, миллион в квадрате. Безумное число, построение графа будет работать годы, а кроме того, непонятно, где вы будете его хранить. Если граф будет весить триллион байт, ни в какую оперативную память он не влезет.
А теперь еще вернитесь в 1990 год, и вы поймете, что у вас даже на диск он не поместится, не умели тогда столько хранить. И даже ни на какой кластер не влезет. Поэтому для произвольной пары ридов, скорее всего, никакого overlay нет. Один, два или три — несущественно. И таким мы интересоваться не хотим. Мы хотим интересоваться только существенным overlay, в десятки и сотни символов.
Можно использовать такую структуру данных, как суффиксное дерево. Вы можете построить суффиксное дерево на всех ваших строчках, на всех ваших ридах, такое объединенное. И дальше для каждого рида в этом суффиксном дереве найти некоторый путь. Он очень сильно сократит количество строк, с которыми вы найдете overlay.
Когда вы построили такой граф, у каждой вершины будет, условно, от 100 до 1000. Это много, десятки миллиардов ребер, но они уже могут поместиться в память или хотя бы на диск, с ними можно работать.
Найдя этот граф, мы должны его немного упростить. Предлагается выкинуть все транзитивные ребра. Скорее всего, транзитивное ребро означает слабое наложение, а еще был промежуточный рид между двумя, которые слабо наложились. Он хорошо накладывается как с первым, так и с последним. Мы ребро выкинем, граф станет проще, и будет понятно, что делать.
Если такие ребра повыкидывать, то граф уже существенно упрощается. По такому графу, как снизу, в целом уже понятно, что делать. Если бы не неприятность посередине, можно было бы сказать, что мы просто слева направо читаем и получаем наш ответ. Ведь мы мы знаем overlay и мы знаем, какие строчки каждой вершине нашего графа соответствуют, мы можем пойти, начитать и составить строчку.
Такие вещи приходится как-то разрешать. Есть много разных сложных алгоритмов, которые это делают.
Это был набросок идеи одного алгоритма. Есть еще другой, применение к указанной задаче предложил Павел Певзнер в 1989 году. Он руководит лабораторией в Санкт-Петербурге, которая написала свой сборщик геномов, сейчас очень популярный и эффективный, и от него пошла вся плеяда биоинформатиков в Санкт-Петербурге, сделавших институт биоинформатики и много других прекрасных проектов.
Идея была в следующем: применить такую чисто дискретную вещь, как граф де Брейна. Мы смотрим строки, все они одинаковой длины, и если все они накладываются друг на друга с точностью до первого символа первой строки и последнего символа второй строки, то мы говорим, что между ними есть ребро. Это почти полностью совпавшие строки, кроме первого и последнего символа.
Оказывается, если построить такой граф, в нем всегда будет эйлеров путь. Если вы взяли строчку и сказали, что у нас есть k = 4, сказали, что вершина состоит из подстрок, идущих подряд при k= 4, а между подряд идущими есть ребра, то вы получите некоторый граф. Ваша строка как-нибудь сожмется, поскольку могут быть повторы в вашей большой строке. Но, прочитав некий эйлеров путь в вашем графе, вы сможете восстановить исходную строку — не всегда точно, иногда из-за больших повторов они могут куда-нибудь переставиться, с ними может что-нибудь случиться. Но в целом с хорошей точностью вы вашу строку восстановите.
Идея — применить это всё к сборке генома.
Вот более конкретный пример, как по строке и заданному k строится граф де Брейна. Мы взяли строку АААВВВА, взяли k = 2, вот вершины из двух символов, построили на них такой граф переходов.
Дальше предлагается взять все ваши риды, на каждом построить, как и на предыдущем слайде, граф де Брейна, и все их объединить в один большой граф. Если вершине соответствует одна строка, то у вас должна быть одна вершина, хоть она и взялась из разных ридов.
Вы строите такой граф. Дальше понятно: поскольку у вас большая плотность покрытия, будет много одинаковых ребер. Их надо склеить. Печальная новость: у вас есть ошибки, из которых вы будете получать лишние ответвления. Их надо будет как-то искать и удалять в этом графе де Брейна.
Вторая плохая новость: эйлерова пути в этом графе вы, скорее всего, не найдете, и эйлеровым он не будет. Но это решается тем, что задачу нужно немного ослабить. Действительно, мы же ищем контиги, у нас могут быть непересекающиеся куски.
Если мы задачу ослабим, она будет формулироваться иначе. Нам надо будет искать не эйлеров путь. Нам надо будет просто искать наименьшее количество циклов, которые покрывают весь граф с точки зрения количества ребер или, желательно, даже не покрывают, а не пересекаются. Другими словамм, мы просто должны будем разложить наш граф на непересекающиеся циклы.
Я рассказал про сборку генома всё, что хотел. Сборка генома — достаточно сложная и интересная задача, достаточно специальная, там применяются и строковые алгоритмы, и графовые алгоритмы, там можно многое улучшать. Очень призываю вас, если вам интересно, почитать про сборку более подробные лекции, материалы. Я могу подсказать, какие.
Гораздо более популярная задача, но попроще, состоит в том, что надо сделать выравнивание. Идея очень простая: если вы одного человека собрали и теперь отсеквенировали второго человека, то вам не надо второго человека собирать с нуля. На самом деле два человека отличаются друг от друга в смысле нуклеотидов их ДНК на полпроцента — порядок примерно такой. Он может чуть-чуть варьироваться в зависимости от того, из одной популяции происходят люди или из разных. Так что вы можете просто взять и найти, где же встречаются риды вашего нового человека в эталонном геноме, который вы уже собрали. Искать вам надо с отличиями, вы ожидаете, что организм, который вы исследуете, отличается от эталонного. Многие отличия будут просто точечными. Бывают еще большие вставки и удаления. Вам просто нужно аккуратно ваши риды поискать в вашем референсном геноме, задетектировать отличия, описать их, и все будет хорошо.
Собственно то, с чем работает большинство биоинформатиков, — выравнивание. Они делают анализ, например, человека, выравнивают его на референсный геном человека, и дальше уже смотрят на отличия. Их они уже исследуют, и с ними работают те, кто занимается геномной биоинформатикой.

Дальше есть задача коллинга вариантов. Когда вы всё повыравнивали, вам надо найти отличия. Там бывают гетерозиготы, их тоже надо отдельно найти.
Какая здесь математика и программирование? Здесь используется много статистических методов. Кажется, можно было просто взять консенсус или посмотреть на две самые частые буквы, но утверждать, что это плохо работает… нет, не плохо, однако можно сделать лучше. Люди строят разные байесовские модели, классификаторы, которые правильно выполняют коллинг.
Дальше есть сложная задача, про которую очень хорошо знает Федя, — аннотация вариантов. Зачем люди ее делают? Например, для диагностики наследственных заболеваний. Для этого им надо понять, приведет ли вариант к плохим последствиям или нет. И тут никакого универсального решения, к сожалению, не существует. Есть много разных методов. Люди пытаются моделировать, понимать, будет белок работать или нет. Но часто случается так, что белок вроде бы сломался от варианта, а никаких плохих последствий для человека нет. Потому что есть соседний белок, который выполняет точно такую же функцию. Просто чуть хуже стало работать, но глобально ничего не сломалось.
Бывают разные статистические методы, когда люди собирают тысячи и десятки тысяч геномов разных популяций и выясняют, что в такой-то позиции никогда, кроме буквы С, ничего не бывало. Наверное, это что-то значит, и буква С здесь не просто так. Потому что у вас мутации в организме происходят случайно, и если в одной позиции возможны разные буквы, то они будут у вас в популяции в целом, если они ничему не мешают. А если там только одна буква, значит, другие буквы чему-нибудь мешают, что-нибудь портят. Такие люди болеют или что-то еще. Там ее быть не может.
Основная суть в следующем: во-первых, есть построение трехмерной структуры и люди на ее основе пытаются предсказывать, а во-вторых, есть статистика и надо уметь анализировать огромные объемы данных. 10 тыс. геномов человека — это какие-нибудь терабайты. По меркам Яндекса и моим это не очень много, но по меркам людей, которые привыкли работать на одном или пяти серверах, это, наоборот, очень много. И анализировать такие данные сложно.
Дальше есть machine learning. Он пытается все эти методы объединить и для конкретной задачи, которую вы решаете, построить некоторую модель, которая бы лучше всего решала задачу.
Дифэкспрессию пропущу.

Про сложности биоинформатики. Важная часть, на которую хочу обратить внимание, — обработка данных. Важно всё, начиная от момента, когда у вас берут пробу и с ней работают химики, биологи в лаборатории, заканчивая тем, как вы работаете с данными, с ридами. Потому что если вы где-нибудь сделаете что-нибудь не то, поступите неправильно, вы дальше получите данные, про которые вы считаете, что здесь есть замена буквы С на букву А, а на самом деле замены нет. Вы сделаете какие-то выводы, и будет очень грустно, если вы напишите статью, а потом выяснится, что на самом деле речь идет о ложной замене, ее там не было. Вы же из-за ошибки во всем анализе решили, что она там есть. Понятно, что такие вещи перепроверяют, используют Сенгера для перепроверки, но перепроверить можно один конкретный вариант, если вы на нем строите вашу статью.
А если вы пишите статью, которая основана на огромной статистике по огромному количеству вариантов, и в своем анализе где-нибудь ошиблись, будет достаточно печально. Здесь нужно очень хорошее понимание, как все устроено. Кажется, что не зная, как работает выравниватель или коллинг, сложно хорошо работать с такими данными. Просто взяв стандартный тул и выровняв им стандартным образом, вы, наверное, сейчас получите что-нибудь неправильное. Есть надежда на исправления. Как верит Андрей, всё стандартизируют и скажут: с такими данными надо делать так, с такими — так. Будем верить. Но пока всё иначе. Пока надо понимать принципы работы.
Дальше есть интересная важная задача. Отсеквенировали вы ДНК, получили последовательность символов, и что дальше? Чтобы что-нибудь понимать, нам нужно очень хорошо знать, где у ДНК гены, где у нее разные области. И есть отдельная наука, например, РНК секвенируют, чтобы улучшить разметку и понимание, где какие гены, как они сплайсятся, как всё устроено. И пока перечисленное — достаточно ручной труд, хорошей автоматики нет, но есть много разных попыток попробовать построить нейронные сети или сложные классификаторы, которые просто по последовательности сказали бы: я смотрю на последовательность ДНК и понимаю, что это регуляционная область.
Смотрю, а тут ген начался. А тут, наверное, интрон начался, потому что вариативность большая и буковки идут так-то. Но это мечты. Определенные результаты в этой области есть, некоторые вещи люди умеют определять просто по последовательности нуклеотидов, но далеко не все. И задача, состоящая в том, чтобы детектировать гены хорошо, кажется, пока не решается.
Есть еще техническая задача, которая тоже, к сожалению, биологами не всегда решается хорошо. Это именно хранение, эффективная обработка данных и унификация. К сожалению, мир устроен так: каждая лаборатория имеет свой кластер, свой банк данных, и понятно, что держать у себя плеяду системных администраторов, разработчиков, которые будут за всем следить, очень сложно. Во-первых, есть специфика задачи и системный администратор, которого вы наймете, ничего про вашу специфику знать не будет. Во-вторых, найти людей, которые хорошо понимают во всем этом выравнивании, тоже достаточно сложно. Так что есть надежда, что когда-нибудь появятся общие базы, куда всё можно заливать, куда можно приходить, смотреть, искать по конкретному варианту генома. Таких баз уже много, но они часто разнообразные, у каждой лаборатории своя, в своем формате. Будем надеяться, что появится унификация и задачи будут решаться лучше. Другими словами, если вы программист, здесь есть что поделать.
В качестве заключения скажу: если вы хотите выучиться биоинформатике, вы можете начать смотреть лекции, читать что-нибудь. Либо вы можете пойти учиться в специализированное заведение, например во ВШЭ на факультет компьютерных наук. В 2016 году там как раз открылась магистратура, по большому счету, по биоинформатике. Она называется «Анализ данных в биологии и медицине», но если вы откроете курс, то в целом он будет по биоинформатике.
Есть еще школа биоинформатики, которую тоже делают ребята совместно с ФББ. Есть Институт биоинформатики в Питере. В общем, много разных вариантов. Всем спасибо.
Под катом вы найдёте расшифровку лекции Игната Колесниченко — выпускника мехмата МГУ и Школы анализа данных. Сейчас Игнат работает ведущим разработчиком службы технологий распределённых вычислений Яндекса.
Кто здесь программист? Примерно половина аудитории. Мой доклад — для вас.
Цель доклада следующая: я в точности расскажу, как берутся данные и как они устроены. Рассказ будет чуть более глубоким, чем у Андрея. Для этого придется немного напомнить про биологию. Я, по крайней мере, рассчитываю, что программисты не знают биологию.
А дальше попробую рассказать, какие алгоритмически сложные задачи решаются в данной области, чтобы заинтересовать вас, чтобы вы поняли, что это здорово и надо идти в биоинформатику, помогать людям решать сложные задачи. Очевидно, сами биологи с алгоритмическими задачами не справятся.
Я окончил мехмат. Я математик, работаю в Яндексе. Я программист, занимаюсь системой YT, мы делаем внутреннюю инфраструктуру в Яндексе, у нас кластера на тысячи машин. Все данные, которые собирает Яндекс, хранятся в этих кластерах, всё обрабатывается, петабайты данных, в этом я немного понимаю. А еще я сооснователь компании «Эбином». Там я занимался такой же технической задачей. Моя цель была — разобраться, как работают эти алгоритмы, программы, и научить их делать это наиболее автоматизированно, чтобы человек мог залить данные на сайт, ткнуть кнопку, и у него бы развернулся кластер, на нём всё быстренько бы посчиталось, свернулось, и данные бы показались человеку.
Пока я решал эту техническую задачу, мне приходилось разбираться, о каких данных идет речь и как с ними работать. Сегодня я расскажу часть того, что узнал.
На Фейсбуке в анонсе к этому мероприятию было выложено видео Гельфанда. Кто его смотрел? Один человек. Гельфанд смотрит на вас неодобряюще.
Мы будем работать с ДНК, но хочется немного контекста вокруг ДНК, так что мы начнем с клетки. Понятно, что биоинформатика как наука о молекулярной биологии интересуется всеми процессами в клетке, но мы сегодня будем больше разговаривать про техническую сторону, про данные и нас будут интересовать, условно, лишь ядро и близлежащие компоненты.
В ядре содержится ДНК. В нем есть ядрышко. Оно вместе с гистонами, которые показывал Андрей, в некотором смысле отвечает за регуляцию и эпигенетику. Есть вторая важная часть — рибосома, складочки вокруг ядра, так называемый эндоплазматический ретикулум. В нем есть специальные комплексы молекул, называемые рибосомами. Оно будут из ваших кусочков ДНК, которые вы сначала из ДНК вычитаете, делать белки.
Третья часть — хроматин: всё вещество, в котором всё ДНК плавает, находится. Оно упаковано достаточно плотно, но между ним всё равно ещё есть много разных регулирующих молекул. Ну и есть мембрана — защита, питание, то, как клетка общается.
Еще в клетке есть многое другое, другие функции. Про них мы говорить не будем, можете почитать в учебниках.
Теперь про информацию, ДНК и РНК. Есть две основных молекулы: дезоксирибонуклеиновая кислота и рибонуклеиновая кислота. Это молекулы одного типа. Когда-то Уотсоном и Маккриком была открыта структура этой молекулы. Интересная история, можете почитать автобиографию, он очень красиво про всё пишет. Она представляет собой двойную спираль, это популярная картинка, популярное buzzword.
Состоит она из четырех более крупных молекул, комплексов из десятков атомов. Это гуанин, цитозин, аденин и тимин. Причем в РНК вместо тимина будет урацил. Когда вы из ДНК будете делать РНК, вместо буквы «Т» вы получите букву «У». Нам важно знать, что в нашем ДНК содержатся эти четыре буквы. С ними мы и будем работать, по большому счету.
У ДНК есть высшая структура. Это не просто спиралька, которая как-то лежит, абы как скручена. Она закручивается вокруг специальных комплексов — гистонов. Потом гистоны закручиваются в некоторые волокна, из которых уже строится буква Х. Перед вами красивая картинка, которую вы видели в учебниках биологии. Обычно про гистоны не говорят, 15 лет назад не говорили, а показывают букву Х. Она устроена таким образом, ДНК там хитрым образом закручено.
Гистоны отвечают за регуляцию. Когда вы захотите прочитать какую-то РНК с ДНК, у вас может не получиться, потому что оно так закрутилось, так спрятано в этой ДНК, что оттуда не читается. А может, что-то поменяется и оно начнет читаться.
И есть разные белки, которые умеют ко всему этому привязываться, умеют это разворачивать, вытаскивать, чтобы вы прочитали. Вторичная структура очень важна, чтобы понимать, какие участки ДНК, из которых вы потом можете делать РНК белки, вам доступны для чтения.
Что такое белок? Основная жизненная сила клетки, многообразие всего живого есть во многом благодаря белкам. Дело в том, что у белка очень высокая вариативность. Речь идет об огромном классе очень разнообразных молекул, которые участвуют в огромной куче молекул. Они имеют очень разные структуры, что и позволяет всей этой сложной химии происходить внутри клетки.
Что нужно знать про белки? Составной блок — аминокислота. Аминокислот в нашем организме, в нашем мире, 20 штук. Говорят, их немножко больше, но в нашем организме их 20. У них тоже есть определенные латинские буквы. Справа нарисован белок схематически, выписано его начало, указано, какие буквы присутствуют. Я не биолог, не помню, что такое V или M, а биологи сразу могут сказать, что буквы означают.
Структура белка имеет несколько уровней, можно на нее смотреть просто как на последовательность, и когда вы биоинформатик, вы часто именно так и смотрите. Хотя вот Артур интересуется более сложными вторичными и третичными структурами.
Мы будем смотреть на это как на последовательность букв. Вторичная и третичная структуры. Есть такие спиральки, альфа-спирали, они на картинке хорошо видны. Есть еще бета-листы, она умеет в достаточно плоские штуки выстраиваться, и есть специальные штуки, которые собирают эти альфа-спирали и бета-листы.
Дальше то, о чем говорил Андрей, — центральная догма молекулярной биологии. Она состоит в том, что есть трансляция и транскрипция, а есть еще много разных процессов обратных, перпендикулярных, которые тоже задействуют РНК, ДНК и белки в разных направлениях. Но мы посмотрим сегодня только на трансляцию и транскрипцию. Справа есть достаточно полная картинка, описывающая эту вещь в эукариотических клетках, в частности в людях.
Есть у вас ген. В учебниках биологии пишут, что это единица наследственности. Для нас же, когда мы смотрим на ДНК, ген — некоторый не такой простой участок. Не то что он транслируется в РН, а дальше из РНК делается белок. Происходит всё немного не так. Во-первых, в указанном участке есть регионы, интроны, которые вырезаются. А есть экзоны, которые потом и будут кодироваться в ваш белок. Если вы посмотрите на ДНК, на 3 млрд букв, то — вот популярная фраза — важных областей там всего 2%. Только 2% отвечают за кодирование ваших генов. И речь идет именно про экзоны.
Если вы возьмете еще интроны, у вас получится не 2%, а 20%. А оставшиеся 80% — это межгенные области, участвующие, по всей видимости, в какой-то степени в регуляции. Но некоторые действительно низачем не нужны, просто так исторически сложилось.
Происходит следующее: у вас есть некий промоутер перед геном, на этот промоутер садится специальная молекула, белок, который будет считывать и из вашего ДНК делать РНК.
Дальше из РНК преобразование идет другой молекулой, которая из него вырезает ненужные интроны. Вы получаете уже готовую матричную РНК, из которой делаете ваш белок трансляцией. При этом отрезаются 3’ и 5’ — некодируемые концы. Когда из РНК вы делаете белок, на самом деле у вас есть триплет, некоторая последовательность, с которой вы можете начать. Всё, что до неё, просто пропускается. Клетка садится, она это пропускает, и начинает всё делать со стартового кодона.
Я употребил слово кодон. А что такое кодоны? как происходит кодирование? Логика простая. В ДНК есть четыре символа: A, C, T, G. В белке 20 символов, 20 аминокислот. Надо как-то из четырех сделать 20. Понятно, что напрямую не получится, надо их как-то закодировать. С точки зрения ученых, которые занимаются кодированием, устройство очень простое, глупое кодирование, мы бы такое легко придумали. Но с точки зрения биологии выполнить такое было непросто.
У вас читается по три буквы ДНК, и каждый триплет букв кодирует определенную аминокислоту. Триплетов букв 64, и 20 аминокислот можно закодировать 64 вариантами. Кодирование получится избыточным. Есть разные триплеты, которые кодируют одно и то же.
Это всё, что нужно знать на сегодня про трансляцию и транскрипцию.
Андрей много говорил про метагеномику, про разные омики. Ученые последние 50–100 лет очень активно изучают, что происходит в клетке, какие там процессы, какая там химия, а не только биология. И они достаточно сильно преуспели в этом вопросе. Может, у них нет совсем полной картины и понимания, но очень многие вещи они знают.
Есть большие карты преобразования разных веществ в друг друга. В частности, большинство элементов, белки, которые описывают, что вообще в клетке происходит.
Вот маленький кусочек, а есть большая картинка, ученые любят ее рисовать. Перед нами примерная упрощенная схема всего, что делается в нашей клетке.
Важно понимать, что все молекулы более-менее всегда в нашей клетке присутствуют — вопрос в концентрациях. Все время ваше ДНК где-нибудь разворачивается, что-нибудь из нее читается, и то, что читается, во многом зависит от того, какая концентрация у разных белков. И вся жизнь клетки — регуляция. У вас чего-нибудь стало больше, это что-нибудь пошло делать больше другого процесса, он начал больше что-то транслировать, появился новый белок, он поучаствовал в каком-нибудь процессе, сказал, что хватит меня производить, породил новый белок, который стал мешать его производству. Такие зацикленные с обратной связью процессы в огромном количестве присутствуют в клетке и здесь в целом представлены.
Откуда берутся данные — эти A, C, T, G? Андрей говорил, что есть секвенаторы, которые вам их прочитают. Давайте поймем подробнее, как эти приборы устроены и что они прочитают.
Есть классический метод Сенгера, который придумали в конце 70-х, и которых позволил в 1989 году начать проект «Геном человека». К 2001 году проект завершили, смогли собрать последовательность некоего «эталонного» человека, состоящую из 3 млрд букв. Мы разобрали все 23 хромосомы и поняли, какие буквы — A, C, T, G — в каждой хромосоме находятся.
Метод Сенгера хороший и надежный, ученые ему доверяют, но он очень медленный. В 1977 году это был страшная жуть, вы работали с некоторыми радиоактивными растворами и веществами в перчатках, находясь за свинцовой штучкой, что-то куда-то заливали в пробирке. Через три часа после этих упражнений, если вы случайно не туда не вылили что-то, вы получали картинку, из которой могли узнать 200 нуклеотидов вашего ДНК.
Понятно, что процесс быстро автоматизировали, и в ходе автоматизации технологии немного поменялись. К середине 2000-х появилась масса новых технологий, которые позволяют делать секвенирование быстро и дешево и не тратить на каждые 400 символов три часа работы дорогостоящего ученого.
Последняя технология, о которой говорил Андрей, — мининанопоры. Мне кажется, в научном сообществе пока есть некоторое сомнение насчет того, станет ли эта технология мейнстримом или нет. Но она точно открывает для нас новые границы.
Все предыдущие технологии считают достаточно точно, в них ошибок мало, речь идет про проценты и доли процентов. В этой технологии ошибок существенно больше, 10-20%, но вы можете читать большие иды. Кажется, в больших приложениях это действительно необходимо.
Сумеют ли это добить до технологии, в которой тоже будет мало ошибок, — неизвестно. Но если смогут, будет очень круто.
Общая картина про секвенирование, про то, как это происходит. У вас берут пробу, анализ крови, анализ слюны, или залезают специальным пинцетом, достают кусочек костного мозга: очень болезненная страшная процедура. Дальше по пробе делается некоторая химия, которая все лишнее удаляет и оставляет, условно, только ваше ДНК, которое в пробе присутствовало.
Дальше вы делаете фрагментацию ДНК, получаете маленькие фрагменты, которые вы дальше размножаете, чтобы их было побольше. Это нужно во многих технологиях. Затем кормите секвенатор, он всё прочитывает некоторым способом.
На выходе — всегда одинаковый fastq-файл. Все приборы работают по-разному, у них разные длины, разные типы ошибок, и если вы будете работать с этими данными, то важно понимать, какие здесь есть типы ошибок, как с такими данными правильно работать. Если вы будете применять один и тот же метод ко всему, вы будете получать грязные данные, на которые нельзя положиться.
Кратко про метод Сенгера. Идея очень красивая. У вас есть ДНК-последовательность, вы ее расплодили, она плавает у вас в пробирке. Дальше вы хотите понять, из чего она состоит. Туда скармливаются специальные белки. Может, там даже будет плавать вся ДНК, но будет плавать и молекула, которая умеет ее разбирать.
У ученых очень простой способ. Как они такие вещи с ДНК научились делать? Они посмотрели, как то же самое делает природа в наших клетках, поняли, какие белки что делают, достали их, и теперь сами могут реплицировать ДНК. Просто берут белки, которые занимаются репликацией, и кидают их в пробирку. Там все само происходит.
Они кидают эти белки, и еще кидают наши ACTG. Среди них есть специальные помеченные, которые, когда она приклеилась при чтении к нашей ДНК, дальше не позволяет клеить еще. Накидав все это дело, взболтнув и дав немного постоять, мы получаем кусочки недочитанной исходной ДНК, которой мы интересуемся. У нас случайным образом что-нибудь приклеивается, и надежда в том, что для каждой длины у нас что-нибудь наклеится, вот в этот момент останавливающая ТТР наклеится и всё нам остановит.
Ещё специальные помеченные ACTG, флуоресцентные. Если на них посветить специальным образом, они будут зелеными, синими, красными или желтыми. Затем все это бросают в гель, там они по массе распределяются, их подсвечивают и получают картинку, как справа. Дальше по ней можно посчитать сверху вниз: более короткие выше, более тяжелые ниже.
В итоге мы получаем fastq-файл. Как он выглядит, нарисовано справа. Это такой текстовый формат, что уже должно программистов вводить в недоумение — зачем хранить в текстовом виде, когда можно сжимать? Вот так сложилось. По факту хранят в таком файлике, просто жмут его каким-нибудь KZIP. В файлике записаны данные в четырех строчках: сначала идет ID вашего одного рида, прочтения. Дальше буквы, которые там прочитались. Потом некоторая техническая строка, и в четвертой строке идет качество. Все приборы умеют выдавать некоторые показатели того, насколько они уверены в прочитанном. Дальше, когда вы будете работать с данными, это надо учитывать.
Суть работы большинства алгоритмов в том, что мы читаем много-много раз с большим покрытием случайные кусочки. Дальше по ним хотим понять, как выглядела исходная ДНК. Понятно, что когда люди начинали проект «Геном человека», никаких других собранных геномов у них толком не было. Бактерии тогда уже научились собирать. И это была страшная задача. У вас есть геном человека, в нем 3 млрд символов. Вы получили кучу кусочков длины 300-400, и по ним надо теперь понять, какая же была последовательность, из которой такие кусочки могли получиться. Выглядит страшно.
Поговорим про алгоритмы. Какие задачи решаются с помощью NGS и как они решаются с алгоритмической точки зрения?
Первая задача — сборка генома. Про нее можно прочитать полугодовой курс лекций, там очень много материалов. Я набросаю только общие идеи, чтобы вы поняли, какие алгоритмы здесь применяются, почему это сложно и интересно.
В чем состоит задача сборки генома? Даны риды в формате fastq, их миллионы или десятки миллионов. Например, вы собираете одну хромосому. А надо вам получить геном или хромосому, которую вы хотите собрать.
Теперь, с программистской точки зрения, как формулируется это задача? У вас есть короткие строки из символов ACTG, на них есть некоторые показатели их качества, веса. И когда вы их собираете, есть еще ограничения на ответ. Когда биологи собирали геном человека, они знали, что на 11 хромосоме есть такие-то гены: мы их в результате долгой работы исследовали и другими способами выяснили, что они там есть. И это хочется учитывать. Было бы глупо собрать что-нибудь, а потом выяснить — того, в чем мы уверены, почему-то нет, поскольку алгоритм так решил.
Надо получить некую суперстроку над всеми короткими строками. Причем мы хотим, чтобы она была как можно меньше. Иначе мы можем просто склеить все наши фрагменты и получить что-то, что никому не годится. Поскольку все строки на входе были получены из нашей хромосомы, значит, надо найти эту хромосому. Если мы делали это с достаточно большим покрытием — условно, с каждым сдвигом есть какая-то строчка — и мы соберем самую короткую, это и будет наш геном. Сказанное не всегда правда, есть разные причины, почему можно что-то потерять или упустить в такой формулировке. Но это достаточное приближение, которое всех устраивает.
Упрощенный пример задачи. Можно собрать суперстроку, которая будет короче, чем их длина 16, и она может выглядеть вот так. Мы взяли последнюю строку ССТА, к ней приклеили предпоследнюю, буквы ТА у них перекрылись. Мы, таким образом, два символа сэкономили. Потом к АС приклеили первую строчку. Вторая строчка TGAC оказалась неудачной, ее пришлось просто так приклеить. Понятно, что пример искусственный. По факту таких приклеиваний в настоящем мире у вас не будет.
Есть плохие новости для вас. Те, кто работает программистом и разбирается в алгоритмах, могут открыть стандартный учебник Гэри Джонсона про сложность алгоритмов и выяснить, что задача о суперстроке является NP-трудной. Кто не знает, на практике это примерно означает, что никакого полиномиального алгоритма решения задачи ожидать не приходится: все известные алгоритмы имеют экспоненциальное время работы. А это оказывается полный ужас: у вас десятки миллионов строк, и как вы за экспоненциальное время будете что-то делать? 2 в степени 10 млн — такого за время жизни Вселенной не дождешься.
Но есть и хорошая новость. у нас не худший вход для задачи, какой-то из вполне реальных. И для него есть надежда решить задачу достаточно хорошо.
Общий подход к решению в следующем: по факту нам не удастся разом собрать всю хромосому непрерывно. Тому есть разные причины. Кажется, проще их не объяснять, а смириться, что так и будет.
Когда мы начнем собирать, мы соберем достаточно большие кусочки из сотен тысяч символов, может даже из миллионов. Дальше нам придется их склеить во всю хромосому размером 100 млн. Эти непрерывные кусочки, которые получится собрать, называются контигами. Мы потом по разным вторичным данным глобально пытаемся понять, как контиги друг с другом соотносятся, и собираем их scaffold’ом. В нашем случае речь идет о хромосоме, если мы собираем хромосому человека.
Есть два алгоритмических метода. Это был конец 80-х и начало 90-х, золотой век людей, которые занимались строковыми алгоритмами. Они всю свою теорию и науку придумали в конце 60-х и начале 70-х, но никому она было не нужна. Я даже был на докладе человека, который придумал суффиксное дерево. Он был только ученым-аспирантом, в аспирантуре придумал суффиксное дерево, написал статью и потом ушел в бизнес. И рассказывал, как в конце 80-х неожиданно ему стали писать письма биологи и спрашивать, как в вашей статье это и это работает. Он был очень удивлен, очень рад за них. Очень вдохновил этот случай.
Зачем все понадобилось? Один из алгоритмов называется Overlap-Layout-Consensus. Мы хотим построить на наших кусочках overlay-граф, дальше мы хотим этот граф упростить, выкинув из него все ненужное, и дальше мы в целом поймем, как будут устроены наши контиги.
Overlay-граф — следующая вещь. Есть у нас два наших рида, мы хотим для каждой — что уже звучит немного безумно с учетом наших данных — пары наших ридов понять, насколько сильно они перекрываются.
Есть такие риды. Они перекрываются на 5, и красным видно, где они перекрываются.
Если мы построим граф этих перекрытий, дальше можно будет с ним работать, пытаться прочитать по этому графу наш геном. Вопрос, как это делать, — отдельный. Очевидно, сразу встает задача для каждой пары ридов построить overlap. У нас ридов ну пусть даже миллион, миллион в квадрате. Безумное число, построение графа будет работать годы, а кроме того, непонятно, где вы будете его хранить. Если граф будет весить триллион байт, ни в какую оперативную память он не влезет.
А теперь еще вернитесь в 1990 год, и вы поймете, что у вас даже на диск он не поместится, не умели тогда столько хранить. И даже ни на какой кластер не влезет. Поэтому для произвольной пары ридов, скорее всего, никакого overlay нет. Один, два или три — несущественно. И таким мы интересоваться не хотим. Мы хотим интересоваться только существенным overlay, в десятки и сотни символов.
Можно использовать такую структуру данных, как суффиксное дерево. Вы можете построить суффиксное дерево на всех ваших строчках, на всех ваших ридах, такое объединенное. И дальше для каждого рида в этом суффиксном дереве найти некоторый путь. Он очень сильно сократит количество строк, с которыми вы найдете overlay.
Когда вы построили такой граф, у каждой вершины будет, условно, от 100 до 1000. Это много, десятки миллиардов ребер, но они уже могут поместиться в память или хотя бы на диск, с ними можно работать.
Найдя этот граф, мы должны его немного упростить. Предлагается выкинуть все транзитивные ребра. Скорее всего, транзитивное ребро означает слабое наложение, а еще был промежуточный рид между двумя, которые слабо наложились. Он хорошо накладывается как с первым, так и с последним. Мы ребро выкинем, граф станет проще, и будет понятно, что делать.
Если такие ребра повыкидывать, то граф уже существенно упрощается. По такому графу, как снизу, в целом уже понятно, что делать. Если бы не неприятность посередине, можно было бы сказать, что мы просто слева направо читаем и получаем наш ответ. Ведь мы мы знаем overlay и мы знаем, какие строчки каждой вершине нашего графа соответствуют, мы можем пойти, начитать и составить строчку.
Такие вещи приходится как-то разрешать. Есть много разных сложных алгоритмов, которые это делают.
Это был набросок идеи одного алгоритма. Есть еще другой, применение к указанной задаче предложил Павел Певзнер в 1989 году. Он руководит лабораторией в Санкт-Петербурге, которая написала свой сборщик геномов, сейчас очень популярный и эффективный, и от него пошла вся плеяда биоинформатиков в Санкт-Петербурге, сделавших институт биоинформатики и много других прекрасных проектов.
Идея была в следующем: применить такую чисто дискретную вещь, как граф де Брейна. Мы смотрим строки, все они одинаковой длины, и если все они накладываются друг на друга с точностью до первого символа первой строки и последнего символа второй строки, то мы говорим, что между ними есть ребро. Это почти полностью совпавшие строки, кроме первого и последнего символа.
Оказывается, если построить такой граф, в нем всегда будет эйлеров путь. Если вы взяли строчку и сказали, что у нас есть k = 4, сказали, что вершина состоит из подстрок, идущих подряд при k= 4, а между подряд идущими есть ребра, то вы получите некоторый граф. Ваша строка как-нибудь сожмется, поскольку могут быть повторы в вашей большой строке. Но, прочитав некий эйлеров путь в вашем графе, вы сможете восстановить исходную строку — не всегда точно, иногда из-за больших повторов они могут куда-нибудь переставиться, с ними может что-нибудь случиться. Но в целом с хорошей точностью вы вашу строку восстановите.
Идея — применить это всё к сборке генома.
Вот более конкретный пример, как по строке и заданному k строится граф де Брейна. Мы взяли строку АААВВВА, взяли k = 2, вот вершины из двух символов, построили на них такой граф переходов.
Дальше предлагается взять все ваши риды, на каждом построить, как и на предыдущем слайде, граф де Брейна, и все их объединить в один большой граф. Если вершине соответствует одна строка, то у вас должна быть одна вершина, хоть она и взялась из разных ридов.
Вы строите такой граф. Дальше понятно: поскольку у вас большая плотность покрытия, будет много одинаковых ребер. Их надо склеить. Печальная новость: у вас есть ошибки, из которых вы будете получать лишние ответвления. Их надо будет как-то искать и удалять в этом графе де Брейна.
Вторая плохая новость: эйлерова пути в этом графе вы, скорее всего, не найдете, и эйлеровым он не будет. Но это решается тем, что задачу нужно немного ослабить. Действительно, мы же ищем контиги, у нас могут быть непересекающиеся куски.
Если мы задачу ослабим, она будет формулироваться иначе. Нам надо будет искать не эйлеров путь. Нам надо будет просто искать наименьшее количество циклов, которые покрывают весь граф с точки зрения количества ребер или, желательно, даже не покрывают, а не пересекаются. Другими словамм, мы просто должны будем разложить наш граф на непересекающиеся циклы.
Я рассказал про сборку генома всё, что хотел. Сборка генома — достаточно сложная и интересная задача, достаточно специальная, там применяются и строковые алгоритмы, и графовые алгоритмы, там можно многое улучшать. Очень призываю вас, если вам интересно, почитать про сборку более подробные лекции, материалы. Я могу подсказать, какие.
Гораздо более популярная задача, но попроще, состоит в том, что надо сделать выравнивание. Идея очень простая: если вы одного человека собрали и теперь отсеквенировали второго человека, то вам не надо второго человека собирать с нуля. На самом деле два человека отличаются друг от друга в смысле нуклеотидов их ДНК на полпроцента — порядок примерно такой. Он может чуть-чуть варьироваться в зависимости от того, из одной популяции происходят люди или из разных. Так что вы можете просто взять и найти, где же встречаются риды вашего нового человека в эталонном геноме, который вы уже собрали. Искать вам надо с отличиями, вы ожидаете, что организм, который вы исследуете, отличается от эталонного. Многие отличия будут просто точечными. Бывают еще большие вставки и удаления. Вам просто нужно аккуратно ваши риды поискать в вашем референсном геноме, задетектировать отличия, описать их, и все будет хорошо.
Собственно то, с чем работает большинство биоинформатиков, — выравнивание. Они делают анализ, например, человека, выравнивают его на референсный геном человека, и дальше уже смотрят на отличия. Их они уже исследуют, и с ними работают те, кто занимается геномной биоинформатикой.
Дальше есть задача коллинга вариантов. Когда вы всё повыравнивали, вам надо найти отличия. Там бывают гетерозиготы, их тоже надо отдельно найти.
Какая здесь математика и программирование? Здесь используется много статистических методов. Кажется, можно было просто взять консенсус или посмотреть на две самые частые буквы, но утверждать, что это плохо работает… нет, не плохо, однако можно сделать лучше. Люди строят разные байесовские модели, классификаторы, которые правильно выполняют коллинг.
Дальше есть сложная задача, про которую очень хорошо знает Федя, — аннотация вариантов. Зачем люди ее делают? Например, для диагностики наследственных заболеваний. Для этого им надо понять, приведет ли вариант к плохим последствиям или нет. И тут никакого универсального решения, к сожалению, не существует. Есть много разных методов. Люди пытаются моделировать, понимать, будет белок работать или нет. Но часто случается так, что белок вроде бы сломался от варианта, а никаких плохих последствий для человека нет. Потому что есть соседний белок, который выполняет точно такую же функцию. Просто чуть хуже стало работать, но глобально ничего не сломалось.
Бывают разные статистические методы, когда люди собирают тысячи и десятки тысяч геномов разных популяций и выясняют, что в такой-то позиции никогда, кроме буквы С, ничего не бывало. Наверное, это что-то значит, и буква С здесь не просто так. Потому что у вас мутации в организме происходят случайно, и если в одной позиции возможны разные буквы, то они будут у вас в популяции в целом, если они ничему не мешают. А если там только одна буква, значит, другие буквы чему-нибудь мешают, что-нибудь портят. Такие люди болеют или что-то еще. Там ее быть не может.
Основная суть в следующем: во-первых, есть построение трехмерной структуры и люди на ее основе пытаются предсказывать, а во-вторых, есть статистика и надо уметь анализировать огромные объемы данных. 10 тыс. геномов человека — это какие-нибудь терабайты. По меркам Яндекса и моим это не очень много, но по меркам людей, которые привыкли работать на одном или пяти серверах, это, наоборот, очень много. И анализировать такие данные сложно.
Дальше есть machine learning. Он пытается все эти методы объединить и для конкретной задачи, которую вы решаете, построить некоторую модель, которая бы лучше всего решала задачу.
Дифэкспрессию пропущу.
Про сложности биоинформатики. Важная часть, на которую хочу обратить внимание, — обработка данных. Важно всё, начиная от момента, когда у вас берут пробу и с ней работают химики, биологи в лаборатории, заканчивая тем, как вы работаете с данными, с ридами. Потому что если вы где-нибудь сделаете что-нибудь не то, поступите неправильно, вы дальше получите данные, про которые вы считаете, что здесь есть замена буквы С на букву А, а на самом деле замены нет. Вы сделаете какие-то выводы, и будет очень грустно, если вы напишите статью, а потом выяснится, что на самом деле речь идет о ложной замене, ее там не было. Вы же из-за ошибки во всем анализе решили, что она там есть. Понятно, что такие вещи перепроверяют, используют Сенгера для перепроверки, но перепроверить можно один конкретный вариант, если вы на нем строите вашу статью.
А если вы пишите статью, которая основана на огромной статистике по огромному количеству вариантов, и в своем анализе где-нибудь ошиблись, будет достаточно печально. Здесь нужно очень хорошее понимание, как все устроено. Кажется, что не зная, как работает выравниватель или коллинг, сложно хорошо работать с такими данными. Просто взяв стандартный тул и выровняв им стандартным образом, вы, наверное, сейчас получите что-нибудь неправильное. Есть надежда на исправления. Как верит Андрей, всё стандартизируют и скажут: с такими данными надо делать так, с такими — так. Будем верить. Но пока всё иначе. Пока надо понимать принципы работы.
Дальше есть интересная важная задача. Отсеквенировали вы ДНК, получили последовательность символов, и что дальше? Чтобы что-нибудь понимать, нам нужно очень хорошо знать, где у ДНК гены, где у нее разные области. И есть отдельная наука, например, РНК секвенируют, чтобы улучшить разметку и понимание, где какие гены, как они сплайсятся, как всё устроено. И пока перечисленное — достаточно ручной труд, хорошей автоматики нет, но есть много разных попыток попробовать построить нейронные сети или сложные классификаторы, которые просто по последовательности сказали бы: я смотрю на последовательность ДНК и понимаю, что это регуляционная область.
Смотрю, а тут ген начался. А тут, наверное, интрон начался, потому что вариативность большая и буковки идут так-то. Но это мечты. Определенные результаты в этой области есть, некоторые вещи люди умеют определять просто по последовательности нуклеотидов, но далеко не все. И задача, состоящая в том, чтобы детектировать гены хорошо, кажется, пока не решается.
Есть еще техническая задача, которая тоже, к сожалению, биологами не всегда решается хорошо. Это именно хранение, эффективная обработка данных и унификация. К сожалению, мир устроен так: каждая лаборатория имеет свой кластер, свой банк данных, и понятно, что держать у себя плеяду системных администраторов, разработчиков, которые будут за всем следить, очень сложно. Во-первых, есть специфика задачи и системный администратор, которого вы наймете, ничего про вашу специфику знать не будет. Во-вторых, найти людей, которые хорошо понимают во всем этом выравнивании, тоже достаточно сложно. Так что есть надежда, что когда-нибудь появятся общие базы, куда всё можно заливать, куда можно приходить, смотреть, искать по конкретному варианту генома. Таких баз уже много, но они часто разнообразные, у каждой лаборатории своя, в своем формате. Будем надеяться, что появится унификация и задачи будут решаться лучше. Другими словами, если вы программист, здесь есть что поделать.
В качестве заключения скажу: если вы хотите выучиться биоинформатике, вы можете начать смотреть лекции, читать что-нибудь. Либо вы можете пойти учиться в специализированное заведение, например во ВШЭ на факультет компьютерных наук. В 2016 году там как раз открылась магистратура, по большому счету, по биоинформатике. Она называется «Анализ данных в биологии и медицине», но если вы откроете курс, то в целом он будет по биоинформатике.
Есть еще школа биоинформатики, которую тоже делают ребята совместно с ФББ. Есть Институт биоинформатики в Питере. В общем, много разных вариантов. Всем спасибо.
Поделиться с друзьями