
Этот метод производит самые точные измерения, которые только можно представить, записывая изменения тока у различных участков ДНК.

Помните сцену из фильма «Матрица», в которой Нео раскрывает всю свою силу, после чего мир предстаёт перед ним в виде бегущих в разные стороны строк кода? Что если бы вы видели окружающий мир таким образом, так что сидящий рядом человек был бы подобен веб-странице, на которую можно щёлкнуть правой кнопкой мыши и изучить элемент и исходный код под ним?
Мы ещё не достигли такого уровня, но недавние достижения в нанопоровом секвенировании, обусловленные разработками программного обеспечения с открытым исходным кодом, позволили существенно сократить время декодирования генома с 15 дней до трёх или даже меньше. Не так давно расшифровка занимала годы! Чтобы понять принцип, лежащий в основе метода, названного UNCALLED [GitHub], мы связались с профессором Майклом Шатцем — обладателем звания BDPs (Почётный профессор Блумберг, Bloomberg Distinguished Professorships) и доцентом кафедры компьютерных наук и биологии в Университете Джона Хопкинса.
Начнём с нанопорового секвенатора. Как рассказал Шатц, «идея его создания возникла ещё около 30 лет назад, и легенда гласит, что первая диаграмма была нарисована на салфетке». В реальности черновик изначального концепта для нанопорового секвенатора был сформирован доктором Дэвидом Димером в блокноте стенографиста шариковой ручкой с красными чернилами!

Представьте себе отверстие настолько крошечное, что в него за раз может пройти только одна-единственная нить ДНК. Протолкните через эту пору ваш генетический материал, и вы получите последовательности А, G, C, T (нуклеотиды аденин, гуанин, цитозин и тимин соответственно), составляющие человеческий геном. Итак, как различать между собой эти четыре строительных блока ДНК?

Как объяснил Шатц, «этот процесс включает самые тонкие измерения, которые только можно представить — измерение разницы в токе между различными участками ДНК. Это происходит на уровне пикоампер (пА, 10−12 А), и мы можем получать такие показания в режиме реального времени». Ещё пять лет назад оборудование, необходимое для этой работы, было доступно только серьёзным исследовательским учреждениям. Сегодня примерно за тысячу долларов вы можете приобрести нанопоровый секвенатор в качестве периферийного устройства, подключающегося к любому компьютеру через USB.
В данных, получаемых в результате секвенирования, обычно присутствует много шума. Но Шатц и его команда, вдохновившись моделью Маркова, разработали нечёткую логику (fuzzy logic) для декодирования каждого белка практически в режиме реального времени. «Это же практически как в “Звёздном пути”, не правда ли? — Рассказал Шатц. — Нуклеотиды проходят через это крошечное отверстие, и мы измеряем ток четыре тысячи раз в секунду». Программное обеспечение декодирует последовательности в режиме реального времени, благодаря чему их можно соотнести с различными маркерами. Например, вы можете определить патогенную бактерию или ген, ассоциированный с раком. Что более важно — вы можете игнорировать ненужные на данный момент фрагменты ДНК.
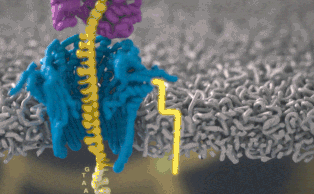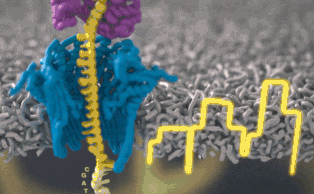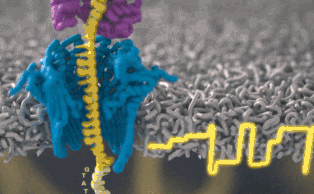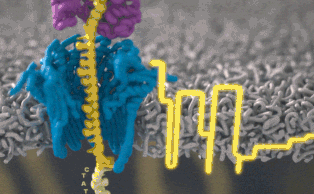
Каждый участок ДНК, проходящий через это крошечное отверстие, представляет собой заряженную молекулу. Софт позволяет пользователю обращать напряжение у каждой такой молекулы, что приводит к её выталкиванию из нанопоры. Именно возможность выборочно секвенировать только те участки, что имеют отношение к научной работе, позволяют добиться значительного повышения скорости секвенирования. Как говорит Шатц: «Существуют API call для выбора молекул, с которыми вы хотите работать. Поразительно, что это вообще возможно».
Обработка «языка жизни»
Каждый фрагмент ДНК возвращает показатели напряжения в зависимости от нуклеотидов. Итак, когда вы получаете напряжение, насколько сложен поисковой запрос? Это не простая таблица, а скорее некое очень нечёткое логическое соответствие. «В электрических данных, которые вам могут понадобиться, требуется, чтобы у нуклеотидов А были одни показатели тока, у С — другие, и так далее, — сказал Шатц. — Но у вас их вообще нет».На самом деле электрический ток связан с несколькими нуклеотидами подряд. Около шести ближайших к считыванию нуклеотидов — самые влиятельные. Прохождение цепочки ДНК через нанопору можно представить как протягивание цепи через трещёточный механизм, по звукам которого можно понять, что через него проходит. «Таким образом, вы на самом деле считываете один и тот же нуклеотид примерно шесть раз в различных контекстах в окружении шести других нуклеотидов». Показатели тока становятся очень шумными. Для каждого измерения тока могут быть сотни последовательностей нуклеотидов, которые он может представлять.
Представьте себе, что каждая комбинация этих шести нуклеотидов обладает смещением. На смещении один имеется сотня возможных последовательностей нуклеотидов, на смещении 2 — ещё одна сотня, на третьем и четвёртом также ещё по сотне последовательностей. «Но это та самая комбинация перекрывающихся последовательностей, в случае с которой вы можете надеяться на решение задачи с конкретными нуклеотидами — ведь вы знаете, что последовательности перекрываются». Например, GATTACA при смещении один может следовать за ATTACAT при смещении два, но не TTTACAT, AATACAT, или любая другая последовательность, начинающаяся не с ATTACA.
Система декодирования использует логику, похожую на обработку естественного языка, чтобы сопоставить шумный электрический сигнал с последовательностями нуклеотидов.
Как только вы получите последовательность нуклеотидов, вам нужно будет обработать текст, чтобы решить, из какой части генома происходит эта молекула. Как говорит Шатц, «основная часть этой технологии была изобретена для систем хранения баз данных ещё 30 лет назад. Существует действительно мощный алгоритм, называемый Преобразование Барроуза — Уилера, который и в наши дни занимает центральное место в геномике».
Нанопоровый секвенатор очень дешёв по сравнению с лабораторными инструментами, используемыми несколько лет назад. Но для секвенирования требуется одноразовый картридж — так называемая проточная ячейка, чья стоимость может увеличиваться при необходимости просмотреть более объёмные последовательности. «Программное обеспечение полезно тем, что вместо сканирования всего генома мы можем действительно точно знать, какую молекулу мы собираемся помещать в наше секвенирование, — сказал Шатц. — Мы можем вытаскивать и выбирать в режиме реального времени, какие молекулы будут полностью считываться, а какие — отбрасываться примерно через секунду секвенирования».

Так, например, если бы вы планировали определить, является ли индивид носителем гена, известного своей связью с раком (например, BRCA1, ассоциированный с раком груди), вы бы взяли образец. Если бы вы хотели обработать весь материал методом нанопорового секвенирования, это был бы очень медленный и дорогостоящий процесс. Все молекулы смешиваются в пробирке, и вы упорядочиваете их по одной за раз, когда они в случайном порядке извлекаются из этой пробирки. Однако новое программное обеспечение из лаборатории Шатца, названное UNCALLED, может практически в реальном времени оценить, стоит изучать последовательность или нет.
Фактически, во время обычного секвенирования, вы захотите секвенировать геном более одного раза, поскольку любой взятый вами образец содержит случайный набор молекул ДНК и может не содержать тех частей, которые вас больше всего интересуют. Имея возможность выбирать, вы можете быстрее отделить то, что вы ищете, и избежать многократного повторения секвенирования в других областях.
Или, например, возьмём пример с инфекционным заболеванием, которое в наши дни у всех на уме. Из-за необходимости в проведении тестирования лаборатории по всему миру сталкиваются с огромными рабочими нагрузками. «В этом смысле человеческий геном невероятно скучный. Это не то, что вы в действительности ищете», — сказал Шатц. С UNCALLED нанопоры будут отсеивать весь явно человеческий материал. «Всё, что не совпадает с человеческим геномом, мы вернём и попробуем сохранить, чтобы провести анализ в режиме реального времени и определить, что это такое».
Открытый доступ к нашему исходному коду
Когда Шатц впервые попал в мир геномики, эта сфера имела крайне плохую репутацию из-за закрытости и проприетарности. «В самом начале было много попыток запатентовать гены. Например, было несколько громких случаев, касающихся генов, ассоциированных с раком груди. Предпринимались попытки запатентовать эти последовательности и получать очень большие деньги с того, что сейчас доступно в ходе обычного анализа».
Как говорит Шатц, к счастью, за последние годы эта тенденция изменилась в лучшую сторону. «За последние 20 лет произошло несколько скачков в развитии технологий, поэтому есть ощущение крайней необходимости. Несмотря на то, что все эти секвенаторы просто записывают нуклеотидные последовательности, каждая платформа обладает различными свойствами и характеристиками, а также ошибками, связанными с ней. Поэтому существует реальная потребность в разработке программного обеспечения, которое может преодолеть эти различия и наилучшим образом использовать данные с разных платформ».
Почему бы не сделать программное обеспечение проприетарным продуктом? Что ж, скорость здесь играет ключевое значение. «Если вы попробуете коммерциализировать это, то для создания компании потребуется некоторое время, настолько много, что к тому моменту, как вы перейдете к работе над продуктом, уже появится следующий. В этой сфере такая гонка, что в долгосрочной перспективе коммерциализировать программное обеспечение очень сложно, указал Шатц. — Кроме того, наша работа в основном финансируется за счёт государственных грантов, поэтому это [бесплатное распространение софта] — один из наших способов вернуть долг обществу».
Текущий климат намного здоровее и доброжелательнее для таких учёных, как Шатц, планирующих продолжать распространять в открытом доступе созданное в его лаборатории программное обеспечение. «Мы получаем так много пользы от возможности делиться кодом и работать сообща. Практически во всех случаях реальные плюсы перевешивают потенциальные минусы».
Комментарии (8)

kuzmin_oleg
28.12.2021 13:53+1Мы ещё не достигли такого уровня, но недавние достижения в нанопоровом секвенировании, обусловленные разработками программного обеспечения с открытым исходным кодом, позволили существенно сократить время декодирования генома с 15 дней до трёх или даже меньше. Не так давно расшифровка занимала годы!
Автор, позвольте с вами не согласиться. Так называемая "расшифровка" действительно долгий процесс и никакое нанопоровое секвенирование не способно значительно ускорить этот процесс. Подход, описанный в статье является рутинным выравниванием ридов (последовательностей буковок, полученных на приборе) на референсную последовательность генома человека. Что же касается именно "расшифровки", то в данном случае процесс растягивается на многие годы. Дело в том, что для организмов, не имеющих готовой сборки референсного генома необходимо не только провести полногеномное секвенирование (это не так долго, сложно, да и не очень дорого), но и помучиться со сборкой этого генома. И не важно, длинные ли используются прочтения (Oxford Nanopore или PacBio), короткие ли (Illumina или IonTorrent), сборка генома занимает не один месяц, а нередко и не один год. А после сборки нужно еще провести аннотирование этого генома, чтобы было понятно где какие гены, за что они отвечают. Аннотирование можно проводить in silico, но все обнаруженные гены подтверждаются по протеому. И вот тут процесс затягивается на многие годы.
Поэтому выравнивание на референс - это быстро (данные секвенирования полного генома человека можно выровнять на референс за пол часа с помощью Illumina Dragen), а вот "расшифровка" генома - это годы.
По остальным спорным пунктам в статье можно еще очень много написать, но это уже будет отдельная статья, а не комментарий.
Den_Che
29.12.2021 08:35Напишите, пожалуйста, особенно интересны детали почему после полногеномного секвенирования сборка неизвестного днк занимает до сих пор "не один месяц". Там есть принципиальные технические трудности или это просто не настолько коммерчески востребованная задача в сравнении с секвенированием отдельных участков днк?
kuzmin_oleg
29.12.2021 11:22В процессе есть как технические трудности, так и определенные проблемы с финансированием. Дело в том, что задача по сборке генома какого-нибудь недавно открытого червячка не настолько коммерчески интересна, как работа с геномом человека или модельных организмов (животных, активно используемых в лабораторной практике для проведения различного рода экспериментов; это, например, мыши, крысы, макак-резус и другие). Хотя даже у модельных организмов не всё так хорошо исследовано и хватает белых пятен. Поэтому для большинства проектов по сборке генома нужно умудриться выбить достаточное финансирование, нипример гранты.
Если рассматривать техническую сторону сборки, то тут тоже много нюансов. В первую очередь нужно использовать прочтения хорошего качества, то есть данные, которым можно доверять. Если кратко, то независимо от используемой технологии секвенирования необходимо перевести "сырые" сигналы, полученные прибором, в так называемый FASTQ-файл (не буду углубляться и расписывать про uBAM). Данный процесс называется Basecalling. FASTQ-файл представляет собой обычный текстовый файл, каждому прочтению соответствует 4 строки этого файла:
@a5d527eb-93d5-456e-95f0-d44b567ab7ce runid=c6bc4820444e313180cdaf567fd11c9b79d03635 read=24 ch=230 start_time=2021-10-20T01:11:25Z flow_cell_id=FAQ14567 barcode=barcode01 barcode_alias=barcode01 GGTATGCACTTCGTTCCAGTTGTATTGCTAAAGGTTAAACTTGCAGACACCGACAACTTTCTTCAACACCTAGACAAGGCAGCAGCACAGGAGGAGCAGGGCGAAGTCCCAGAACCCCAAGTGCTGGCTCTCGGGGTCTCCAGGCCCCGAAGGCGGTGTATGGATTGGGGCCAGCGTTGGGAGGATTCATCTCACCGGTTCTCTTCTTACTGACAGCTGGTGGTGTCCTTCTTCGAAATACTCACGACGCGGGACCCGTTCTCTCCCATTGGGTGTCGGGTTTCTAGAGTCGCCAATCAGCGTCGCCGGGGTCCCGGTTCTAAAGTCCCCACTCACCAACCGGACAAGGTCTCCGCAGACGCCGAGGATGGCAATCATGGCGCTGAGCCCTCCTGGTGCTCTCAGGGGTCCTGGCCTGACCCAGACCCGGGCGGGTGAGTGCGGGGTCGCGAGGAAACGGCCTCTGCCGGAGCAGCGAGGGGCCCGCCCGGCGGGGCGCAGGACCGGGGGAGCCGCGCGGGGAGGAGGGTCGGGCGGGTCTCAGCCTCTCCTCGCCCCCGAGCGCCTCCACTCCATGAGGTATTTCTACACCCGCCGTGTCCCGGCCCGGCCGCTGGGGCCCCGCTTCATCGCCGTGGGCTACGTGGACGACACGCAGTTCGTGCGGTTCGACAGCGACGCCGAGGAGTCCGAGGATGG + '&&&'&(%%%'$#%(2()..0*$%&)*)((&&')&.,++*(%$$$&(&'/)(,&),,,742=AA0(({;36.+/*%'()')''%%%2{47<?{.2,,))674+&%%$,.-/,(&&(537+,+10/.,,.;{:''+146;{'(*,-241/.32;:58;{=6159:66%$&%%%)''*55)&,1,-(&&'$%&,*%&&$$&+2'{{=;3,+*+34)'$%%%$%'()()+))+*((&#&21..---((*+216*).7422*'(*3+(;:<<<<>=86{@;+'()1495,,3{27;=<=777535876334525>:5620+002'))24336:<948=...1++++*/,#%&)%54{+'''-00'((..049/'&&%%$%#"#%&&(%$%(&+30/&)2.0186{1>+,:>@76/-.088;=<663:;<;-(**/9412229:667403;88190-.11975-./:::422,-%'%'*-/*(+41.*7/616.(()-+17,{<10<<851''45/++:A?{{0*,*.0017<>;6149:10A>;;<...8<?{{7578C?=74'&)$')&)'+-,,//0////3$#$'''/673{35<<<>?{:D=865781:68>+++327,'.--076/;:>A@@=B{BA:8<777::8:7455255;9;;>?=:;B8:;?=0+*+-01101;:;66),/./((32::41-Привёл пример одного рида (прочтения) из данных нанопорового секвенирования. В первой строке записывается ID прочтения и дополнительная информация, которая может быть полезна в дальнейшей работе. Во второй строке записаны обнаруженные буковки. В третьей строке записывается направление прочтения знаками "+" или "-", в случае нанопора увидим только "+". А четвертая строка самая интересная. Тут записано качество секвенирования, уверенность прибора в качестве распознавания каждого отдельного нуклеотида. Это так называемый phred quality score, закодированный с помощью одного из символов ASCII, потому что необходимо чтобы одно- или двузначное значение качества соответствовало одному нуклеотиду.
!"#$%&'()*+,-./0123456789:;<=>?@ABCDEFGHI Символ ASCII | | | | | | | | | 0....5...10...15...20...25...30...35...40 Phred score (Q) | | | | | | | | | хуже................................лучшеТак вот, возвращаясь к качеству ридов, то у нанопора всё довольно печально, зачастую риды имеют качество Q10, или одна ошибка на на 10 нуклеотидов (10%). Это очень низкое качество. Хотя сейчас у компании Oxford Nanopore Technologies вышло обновление химии и нанопор, которое позволяет получать риды с качеством Q20 (1 ошибка на 100 нуклеотидов), что уже значительно лучше для работы. Огромное преимущество нанопорового секвенирования - возможность получать очень длинные прочтения, больше миллиона нуклеотидов. Есть секвенатор, позволяющий получать длинные прочтения высокого качества - PacBio, но у него есть один недостаток: цена на прибор начинается от миллиона долларов, при этом затраты на химию для секвенирования тоже довольно ощутимые. Поэтому далеко не все коллективы могут себе позволить использовать PacBio. Приборы компании Illumina позволяют получать прочтения с качеством Q30+ (одна ошибка на 1000 нуклеотидов), точность секвенирования 99,9%. Но есть одно "но" - риды короткие, в среднем 150-300 букв, зависит от используемой химии.
Тенденции по сборке геномов таковы, что сейчас зачастую используют так называемые гибридные сборки, используя как длинные, но не очень точные прочтения, так и короткие, но точные. Длинные прочтения позволяют получать длинные контиги (по сути, перекрывающиеся риды объединяются в более длинную последовательность), затем, на следующем этапе контиги объединяются в скаффолды, всё это дополнительно полируется короткими точными прочтениями. И вот на руках у биоинформатика есть несколько скаффолдов, которые имеют пропуски внутри (серия из N, означает, что на данный момент не удалось определить какие здесь нуклеотиды, только примерное их количество) К какой хромосоме принадлежат данные скаффолды? Нужны дополнительные эксперименты. Полностью ли покрыта хромосома? Какие там гены? Верно, опять нужны дополнительные эксперименты. Это тянет за собой очень много работы как для "мокрых" биологов, которые проводят эксперименты в лаборатории, так и биоинформатиков, так и значительные денежные вливания, потому что это банально дорого. Плюс ко всему для больших геномов нужны серьезные вычислительные мощности, связано это с приличными объемами данных (.gz архив генома человека со средним покрытием 30х (в среднем, каждая буква генома прочитана 30 раз) весит около 100 ГБ), при этом во время работы промежуточных файлов может быть много больше, в моей практике был один случай, когда дополнительных файлов набралось больше чем на 6 ТБ. Далее, если даже более простые операции с полногеномными данными, например, на сервере с 96 ядрами, 1 ТБ оперативной памяти, и данные лежат на SSD, занимают десятки часов, а иногда и сутки. А сборка генома с нуля (de novo) очень требовательна к ресурсам и идёт днями. При этом легко может быть такая ситуация, что после того, как прошла сборка результаты получились так себе, надо подбирать и оптимизировать параметры, и запускать сборку снова, и снова.
Если кому-то получится создать дешевый и быстрый метод для сборки и расшифровки генома, то биологи вздохнут с облегчением, но до этого пока очень далеко. Как человек, который варится во всей этой кухне, могу сказать, что очень много всего делается "на коленке". Да, для большинства вещей есть вполне себе конкретные процедуры: бери это, добавь то, перемешай, проинкубируй и получишь результат. Или возьми эти данные, прогони по такому-то пайплайну и будет тебе счастье. Но шаг влево-вправо и всё, многие стандартные процедуры не работают, что-то можно адаптировать и оптимизировать, а многое приходится изобретать с нуля.

shadrap
29.12.2021 16:38Есть секвенатор, позволяющий получать длинные прочтения высокого качества - PacBio, но у него есть один недостаток: цена на прибор начинается от миллиона долларов, при этом затраты на химию для секвенирования тоже довольно ощутимые.
Я бы сказал не химия дорогая , там все копейки стоит, а желание производителя ее продавать , делает стоимость реактивов действительной ощутимой))
shadrap
парадокс в том, что возможно этот метод и производит самые точные измерения токов в природе, но по сути своей применимости , является самым не точным)) Увы, нанопоровое секвенирование не может пока похвастаться точностью, 12-15% средний процент ошибок. Несомненно , метод был бы хорош на длинных геномах, где за счет перекрытия ридов можно было бы привести %% ошибок к близкому в NGS, но реальная ДНК это не просто цепочка нуклеотидов , а еще и всякое г.... , типа метил, аденил и тп групп пристающих к ДНК из-за чего длинные цепочки просто иногда застревают в поре.
Хотя конечно метод, где отсутствует длинный химический цикл, очень обещающий для повсеместного применения, можно сказать- домашнего , секвенаторов и прочих анализирующих устройств.
Un_ka
Что делают когда ДНК застревает в нанопоре? Как в неё вообще попадают — это же не нитку в игольное ушко продеть.
kuzmin_oleg
В идеале - ДНК не должна застревать. И обычно пролетает со скоростью около 400 нуклеотидов в секунду. Но если происходит загрязнение нанопоры, то управляющий софт автоматически начинает освобождение нанопоры. Как точно это происходит - информации не находил, может быть производитель об этом и не расскажет. Поскольку нанопоры живые, то в процессе секвенирования они умирают и секвенирование через них не идёт. Могу предположить, что заблокированная нанопора, которая не смогла очиститься также помечается софтом как мёртвая и сигнал с неё не учитывается.
Чтобы молекула ДНК попала в нанопору используется несколько механизмов. 1 - поскольку молекула ДНК имеет заряд, то в электрическом поле движется от катода к аноду. 2. На концы молекул в процессе пробоподготовки пришивают дополнительные элементы, такие как баркоды (последовательности нуклеотидов, которые потом позволяют отличить молекулы одного образца от молекул из другого образца). Дополнительно еще присоединяется специальный моторный белок, который затем прикрепляется к нанопоре и проталкивает молекулу ДНК. Плюс в растворе вокруг плавает фермент хеликаза, который обеспечивает расплетение двуцепочечной молекулы ДНК. Поэтому через нанопору проходит только одна из цепей молекулы. А поскольку таких молекул не одна, а миллионы или миллиарды (зависит от длины молекул), то попадают в нанопору они без проблем.
shadrap
Нанопора это рекомбинантный белок, один из бактериальных трансмембранных белков, одним концом прилепленый к CMOS пластине , а другим к типа ДНК полимеразе, хотя скорее это хеликаза, которая называется в их технологии моторный белок. Она крепится к этому альфа-порину и запускает внутрь поры одноцепочечную ДНК. У мотора и поры высокая афинность - липнут друг к другу.