
Все готово, чтобы рассказать Хабр аудитории о применении FPGA в сфере научных высокопроизводительных вычислений. И о том, как на данной задаче надо удалось значительно обскакать GPU (Nvidia K40) не только в метрике производительность на ватт, но и просто с точки зрения скорости вычисления. В качестве FPGA платформы использовался кристалл Xilinx Virtex-7 2000t, подключенный по PCIe к хост компьютеру. Для создания аппаратного вычислительного ядра использовался язык C++ (Vivado HLS).
Под катом текст нашей оригинальной статьи. Там, как обычно бывает, сначала идет долгое описание зачем это все надо и модели, если нет желания это читать, то можно переходить сразу к реализации, а модель посмотреть потом при необходимости. С другой стороны без хотя бы беглого ознакомления с моделью читатель не сможет получить впечатление о том, какие сложные вычисления можно реализовать на FPGA.
Аннотация
В данной работе рассмотрена аппаратная реализация расчета деполимеризации белковой микротрубочки методом броуновской динамики на кристалле программируемой логической интегральной схемы (FPGA) Xilinx Virtex-7 с использованием высокоуровневого транслятора с языка Си Vivado HLS. Реализация на FPGA сравнивается с параллельными реализациями этого же алгоритма на многоядерном процессоре Intel Xeon и графическом процессоре Nvidia K40 по критериям производительности и энергоэффективности. Алгоритм работает на броуновских временах и поэтому требует большого количества нормально распределенных случайных чисел. Оригинальный последовательный код был оптимизирован под многоядердную архитектуру с помощью OpenMP, для графического процессора — с помощью OpenCL, а реализация на FPGA была получена посредством высокоуровневого транслятора Vivado HLS. В работе показано, что реализация на FPGA быстрее CPU в 17 раз и быстрее GPU в 11 раз. Что касается энергоэффективности (производительности на ватт), FPGA была лучше CPU в 160 раз и лучше GPU в 80 раз. Ускоренное на FPGA приложение было разработано с помощью SDK, включающего готовый проект FPGA, имеющий PCI Express интерфейс для связи с хост-компьютером, и софтверные библиотеки для общения хост-приложения с FPGA ускорителем. От конечного разработчика было необходимо только разработать вычислительное ядро алгоритма на языке Си в среде Vivado HLS, и не требовалось специальных навыков FPGA разработки.
Введение
Высокопроизводительные вычисления проводят на процессорах (CPU), объединенных в кластеры и/или имеющих аппаратные ускорители – графические процессоры на видеокартах (GPU) или программируемые логические интегральные схемы (FPGA) [1]. Современный процессор сам по себе является отличной платформой для высокопроизводительных вычислений. К достоинствам CPU можно отнести многоядерную архитектуру с общей когерентной кэш-памятью, поддержку векторных инструкций, высокую частоту, а также огромный набор программных средств, компиляторов и библиотек, обеспечивающий высокую гибкость программирования. Высокая производительность платформы GPU основывается на возможности запустить тысячи параллельных вычислительных потоков на независимых аппаратных ядрах. Для GPU доступны хорошо зарекомендовавшие себя средства разработки (CUDA, OpenCL), снижающие порог использования GPU платформы для прикладных вычислительных задач. Несмотря на это, в последние годы FPGA все чаще стали использоваться в качестве платформы для ускорения задач, в том числе использующих вещественные вычисления [2]. FPGA обладают уникальным свойством, резко отличающим их от CPU и GPU, а именно возможностью построить конвейерную аппаратную схему под конкретный вычислительный алгоритм. Поэтому, несмотря на значительно меньшую тактовую частоту, на которой работают FPGA (по сравнению с CPU и GPU), на некоторых алгоритмах на FPGA удается добиться большей производительности [3]–[5]. С другой стороны, меньшая частота работы означает меньшее энергопотребление, и FPGA практически всегда более эффективны, чем CPU и GPU, если использовать метрику «производительность на ватт» [5].
Одним из классических приложений, требующих высокопроизводительных вычислений является метод молекулярной динамики, использующийся для расчета движения систем атомов или молекул. В рамках этого метода взаимодействия между атомами и молекулами описываются в рамках законов Ньютоновской механики с помощью потенциалов взаимодействия. Расчет сил взаимодействия проводится итеративно и представляет существенную вычислительную сложность, учитывая большое количество атомов/молекул в системе и большое количество расчетных итераций. Ускорению расчетов молекулярной динамики было уделено много внимания в литературе в различных системах: суперкомпьютерах [6], кластерах [7], специализированных под молекулярно-динамические расчеты машинах [8]–[10], машинах c ускорителями на основе GPU [11] и FPGA [12]–[17]. Было продемонстрировано, что FPGA может являться конкурентной альтернативой в качестве аппаратного ускорителя для молекулярно-динамических вычислений во многих случаях, однако на сегодняшний день не существует консенсуса о том, для каких именно задач и алгоритмов выгоднее применять платформу FPGA. В данной работе мы рассматриваем важный частный случай молекулярной динамики – броуновскую динамику. Основная особенность метода броуновской динамики по сравнению с молекулярной динамикой заключается в том, что, молекулярная система моделируется более грубо, т.е. в качестве элементарных объектов моделирования выступают не отдельные атомы, а более крупные частицы, такие как отдельные домены макромолекул или целые макромолекулы. Молекулы растворителя и другие малые молекулы в явном виде не моделируются, а их эффекты учитываются в виде случайной силы. Таким образом удается значительно снизить размерность системы, что позволяет увеличить интервал времени, покрываемый модельными расчетами на порядки. Нам неизвестны описанные в литературе попытки исследовать эффективность FPGA по сравнению с альтернативными платформами для ускорения задач броуновской динамики. Поэтому мы предприняли исследование данного вопроса на примере задачи моделирования деполимеризации микротрубочки методом броуновской динамики. Микротрубочки – это трубки диаметром около 25 нм и длиной от нескольких десятков нанометров до десятков микрон, состоящие белка тубулина и входящие в состав внутреннего скелета живых клеток. Ключевой особенностью микротрубочек является их динамическая нестабильность, т.е. возможность спонтанно переключаться между фазами полимеризации и деполимеризации [18]. Это поведение важно прежде всего для захвата и перемещения хромосом микротрубочками во время клеточного деления. Кроме того, микротрубочки играют важную роль во внутриклеточном транспорте, движении ресничек и жгутиков и поддержании формы клетки [19].
Механизмы, лежащие в основе работы микротрубочек, исследуются уже несколько десятков лет, но лишь недавно развитие вычислительных технологий позволило описывать поведение микротрубочек на молекулярном уровне. Наиболее подробная молекулярная модель динамики микротрубочек, созданная недавно нашей группой на основе метода броуновской динамики, была реализована базе CPU и позволяла рассчитывать времена полимеризации/деполимеризации микротрубочек порядка нескольких секунд [20]. Это пролило свет на ряд важных аспектов динамики микротрубочек, однако, тем не менее, многие ключевые экспериментально наблюдаемые явления остались за рамками теоретического описания, т.к. они происходят в микротрубочках на временах десятков и даже сотен секунд [21]. Таким образом, для прямого сравнения теории и эксперимента критически важно достигнуть ускорения расчетов динамики микротрубочек хотя бы на порядок.
В данной работе мы исследуем возможность ускорения расчетов броуновской динамики микротрубочки на FPGA и сравниваем результаты, полученные при реализации одного и того же алгоритма динамики микротрубочек на трех разных платформах, по критериям производительности и энергоэффективности.
Математическая модель
Общие сведения о структуре микротрубочки
Структурно микротрубочка представляет собой цилиндр, состоящий из 13 цепочек – протофиламентов.

На рисунке слева – схема модели микротрубочки. Серым показаны субъединицы тубулина, черными точками – центры взаимодействия между ними. Справа — вид энергетических потенциалов взаимодействия между тубулинами.
Каждый протофиламент построен из димеров белка тубулина. Соседние протофиламенты связаны друг с другом боковыми связями и сдвинуты относительно друг друга на расстояние 3/13 длины одного мономера, так что микротрубочка имеет спиральность. При полимеризации димеры тубулина присоединяются к концам протофиламентов, причем протофиламенты микротрубочки стремятся принимать прямую конформацию. При деполимеризации боковые связи между протофиламентами на конце микротрубочки разрываются, и протофиламенты закручиваются наружу. При этом от них случайным образом отрываются олигомеры тубулина.
Моделирование деполимеризации микротрубочки методом броуновской динамики
Используемая здесь молекулярная модель микротрубочки была впервые представлена в статье [20]. Поскольку задачей настоящего исследования являлось сравнение производительности различных вычислительных платформ, мы ограничились моделированием только деполимеризации микротрубочки.
Вкратце, микротрубочка моделировалась как набор сферических частиц, представляющих собой мономеры тубулина. Мономеры могли двигаться только в соответствующей им радиальной плоскости, т.е. в плоскости, проходящей через ось микротрубочки и соответствующий протофиламент. Таким образом, положение и ориентация каждого мономера полностью определялись тремя координатами: двумя декартовыми координатами центра мономера и углом ориентации. Каждый мономер имел четыре центра взаимодействия на своей поверхности: два центра бокового взаимодействия и два центра продольного взаимодействия. Энергия тубулин-тубулинового взаимодействия зависела от расстояния r между сайтами взаимодействия на поверхности соседних субъединиц и от угла наклона между соседними мономерами тубулина в протофиламенте. Боковые и продольные взаимодействия между димерами тубулина определялись потенциалом, имеющим следующий вид:
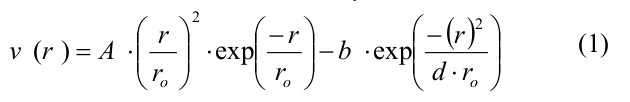
где A и b определяли глубину потенциальной ямы и высоту энергетического барьера, r0 и d – параметры, задающие ширину потенциальной ямы и форму потенциала в целом. Параметр A принимал различные значения для боковых и продольных связей, так что боковые взаимодействия были слабее продольных, все остальные параметры совпадали для обоих типов связей (полный список параметров и их значений представлен в Table 1 в [20]). Продольные взаимодействия внутри димера моделировались как неразрывные пружины с квадратичным энергетическим потенциалом u_r:
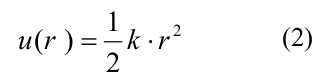
где k — жесткость связи тубулин-тубулинового взаимодействия. Энергия изгиба g(?) связана с поворотом мономеров друг относительно друга и также описывалась квадратичной неразрывной функцией:

где ? — угол между соседними мономерами тубулина в протофиламенте, ? 0 — равновесный угол между двумя мономерами, B — изгибная жесткость. Полная энергия микротрубочки записывалась следующим образом:

где n — номер протофиламента, i — номер мономера в n-ом протофиламенте, Kn — число субъединиц тубулина в n-ом протофиламенте, бокового взаимодействия v_k_lateral — энергия бокового взаимодействия между мономерами, v_k_longitudinal — энергия продольного взаимодействия между димерами.
Эволюция системы рассчитывалось с помощью метода Броуновской динамики [22]. Изначальной конфигурацией микротрубочки была короткая «затравка», содержащая 12 мономеров тубулина в каждом протофиламенте. Мы рассматривали только деполимеризацию МТ и моделировали все тубулины с равновесным углом ? 0 = 0.2 рад. Координаты всех мономеров системы на i-ой итерации выражались следующим образом:
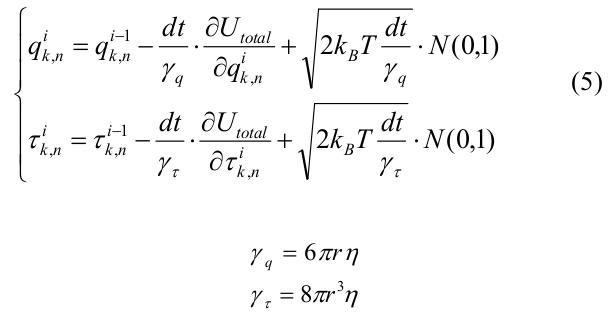
где dt — шаг по времени, U total выражается через (4), k_B — постоянная Больцмана, T- температура, N(0,1) – случайное число из нормального распределения, сгенерированное с помощью алгоритма вихрь Мерсенна [23]. ? q и ? ? — вязкостные коэффициенты сопротивления для сдвига и поворота соответственно, рассчитанные для сфер радиуса r = 2 нм.
Производная полной энергии по независимым координатам q{k,n} выражалась через боковую, продольную составляющие энергии взаимодействия между соседними димерами и внутри димера, а также энергию изгиба:
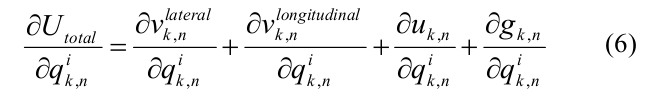
Для ускорения расчетов были использованы аналитические выражения для
всех градиентов энергии:
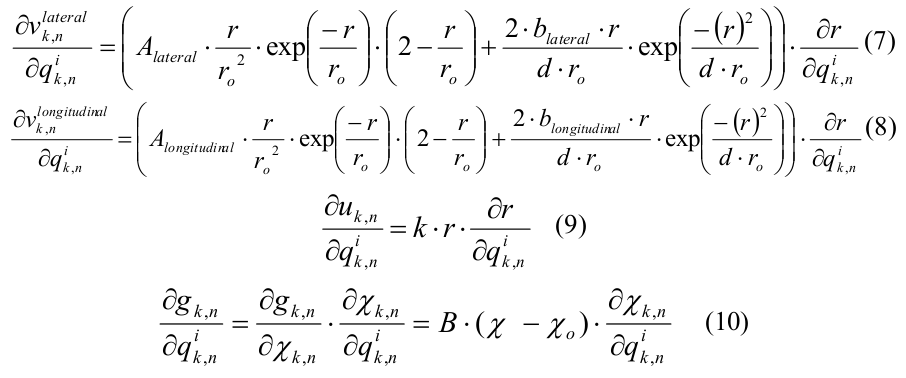
Следует отметить, что размер данной задачи сравнительно мал. Мы рассматривали только 12 слоев мономеров, что дает полное число частиц равное 156. Однако, это нисколько не уменьшает значимость вычислений, т.к. в реальных расчетах достаточно вычислять положение крайних нескольких (порядка 10) слов мономеров, т.к. при росте микротрубочки дальние от конца микротрубочки молекулы тубулина образуют устойчивую цилиндрическую конфигурацию, и брать их в расчет нет смысла.
Псевдокод алгоритма расчета
Алгоритм является итеративным по времени с шагом 0.2 нс. Существуют массив трехмерных координат молекул, а также массивы сил поперечного (латерального) и продольного (лонгитудального) взаимодействий. На каждой итерации по времени последовательно выполняются два вложенных цикла по молекулам, в первом производится вычисление сил взаимодействия по известным координатам, во втором – обновляются сами координаты. В цикле вычисления сил взаимодействия необходимо прочитать координаты трех молекул, одна центральная и две соседние («левая» и «верхняя», см. Рис.2), а результатом вычисления будет сила поперечного взаимодействия между центральной и левой молекулами и сила продольного взаимодействия между центральной и верхней.
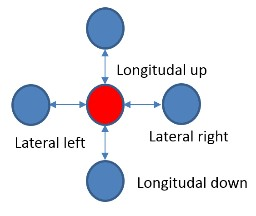
Рис. 2. Схема расположения взаимодействующих субъединиц в модели микротрубочки
В итоге после этого цикла оказываются вычисленными все силы взаимодействия между всеми молекулами. В цикле обновления координат по известным силам вычисляются изменения координат, а также берутся в расчет случайные добавки для учета Броуновского движения. Таким образом, псевдокод алгоритма можно записать следующим образом.
Вход: массив координат молекул M = {x, y, teta}. Граничные условия на силы
взаимодействия.
Выход: массив координат M после K шагов по времени
for t in {0.. K-1} do
for i in {0.. 13} // количество протофиламентов
for j in {0.. 12} do // количество слоев молекул
Mc <- M[i,j]
Ml <- M[i+1,j]
Mu <- M[i,j+1]
// по формулам (7, 8, 9, 10)
F_lat[i,j] <- calc_calteral(Mc, Ml)
F_long[i,j] <- calc_long(Mc, Mu)
end for
end for
for i in {0.. 13}
for j in {0.. 12} do
// по формулам (5)
M[i,j] <- update_coords(F_lat[i,j], F_long[i,j])
end for
end for
end forПрограммная реализация на CPU и GPU
Реализация на CPU
Была предпринята попытка максимально распараллелить код на CPU Intel Xeon E5-2660 2.20GHz под управлением ОС Ubuntu 12.04 с помощью библиотеки OpenMP. Параллельная секция начиналась до цикла по времени. Циклы расчета сил взаимодействия и обновления координат были распараллелены с помощью директивы omp for schedule(static), между циклами была вставлена барьерная синхронизация. Массивы, содержащие силы взаимодействия и координаты молекул, были объявлены как private для каждого потока.
При реализации расчетов на CPU было обнаружено, что размер задачи не позволял ее эффективно распараллелить. Зависимость времени выполнения одной итерации от числа параллельных потоков была немонотонна. Минимальное время расчета одной итерации по времени было получено при использовании всего 2 потоков (ядер CPU). Объясняется это тем что, с увеличением количества потоков растет время на копирование данных между потоками и на их синхронизацию. При этом размер задачи очень мал, чтобы выигрыш от увеличения количества ядер превысил эти накладные расходы. При этом эксперименты показали, что задача слабо масштабируется при увеличении размера (weak scaling), т.е. при дновременном увеличении размера задачи и числа параллельных потоков время вычисления оставалось примерно одинаковым. В итоге лучшим результатом на данном CPU было 22 мкс на одну итерацию по времени при использовании двух ядер CPU. Код не был векторизован из-за сложности вычислений сил взаимодействия.
Реализация на GPU
Мы запускали OpenCL реализацию на граф процессоре Nvidia Tesla K40. Циклы, вычисляющие силы взаимодействия и обновления координат были распараллелены, главный цикл по времени был итеративным. Были реализованы два варианта – с одной и несколькими рабочим группами (work groups). В первом случае было выделено по одному рабочему потоку (work item) на каждую молекулу. В каждом потоке был цикл по времени, в котором вычислялись силы и координаты молекулы потока. При этом применялась барьерная синхронизация после вычисления сил и после обновления координат. В этом случае участие хоста не требовалось для вычислений, он только занимался управлением и запуском ядер.
Во втором случае были два типа потоков, в одном просто вычислялись силы для одной молекулы, во втором – обновлялись координаты. Главный цикл по времени был на хосте, который управлял запуском и синхронизацией ядер на каждой итерации цикла по времени.
Наибольшая производительность была получена в расчетах с одной группой потоков и барьерной синхронизацией между ними. Без использования генераторов псевдослучайных чисел одна итерация вычислялась в течение 5 мкс, если использовать один генератор чисел на все потоки, то время работы возрастало до 9 мкс, а при максимальном заполнении общей памяти (shared memory) удавалось включить 7 независимых генераторов, при этом время вычисления одной итерации по времени составило 14 мкс, что было в 1.57 раза быстрее реализации на CPU.
Загруженность ядер GPU cоставила 7% от одного мультипроцессора (SM), при этом общая память, где размещались массивы сил, координат и буферы данных генераторов псевдослучайных чисел, была заполнена на 100%. Т.е. с одной стороны размер задачи был явно мал для полной загрузки GPU, с другой стороны при увеличении размера задачи пришлось бы использовать глобальную DDR память, что могло бы привести к ограничению роста производительности.
Реализация на FPGA
Описание платформы
Вычисления на FPGA производились на платформе RB-8V7 производства фирмы НПО “Роста”. Она представляет собой 1U блок для установки в стойку. Блок состоит из 8 кристаллов FPGA Xilinx Virtex-7 2000T. Каждая FPGA имеет 1 GB внешней DDR3 памяти и PCI Express x4 2.0 интерфейс к внутреннему PCIe коммутатору. Блок имеет два интерфейса PCIe x4 3.0 к хост-компьютеру через оптические кабели, которые должны быть соединены со специальным адаптером, установленным в хост-компьютер.
В качестве хост-компьютера был использован сервер с CPU Intel Xeon E5-2660 2.20 GHz, работающий под управлением ОС Ubuntu 12.04 LTS – такой же как и для вычислений просто на CPU с помощью OpenMP. Программное обеспечение, работающее на CPU хост-компьютера «видит» блок RB-8V7 как 8 независимых FPGA устройств, подключенных по шине PCI Express. Далее будет описываться взаимодействие CPU только с одной FPGA XC7V72000T, при этом система позволяет использовать FPGA независимо и параллельно.
Ускоренное с помощью FPGA приложение было разработано с помощью SDK со следующей моделью. На CPU хост-компьютера (далее просто CPU) работает основная программа, которая использует ускоритель FPGA для наиболее вычислительно емких процедур. CPU передает данные в ускоритель и обратно через внешнюю DDR память, подключенную к FPGA, а также управляет работой вычислительного ядра в FPGA. Вычислительное ядро создается заранее на языке C/C++, верифицируется и транслируется в RTL код с помощью средства Vivado HLS. RTL код вычислительного ядра вставляется в основной FPGA проект, в котором уже реализована необходимая логика управления и передачи данных, включающая PCI Express ядро, DDR контроллер и шину на кристалле (Рис. 3). Основной FPGA проект иногда называют Board Support Package (BSP), он разрабатывается производителем оборудования, и от пользователя не требуется его модификации. Вычислительное ядро HLS после запуска само обращается в DDR память, считывает оттуда входной буфер данных для обработки и записывает туда же результат вычислений. На уровне языка C++ обращение в память происходит через аргумент функции верхнего уровня вычислительного ядра типа указатель.
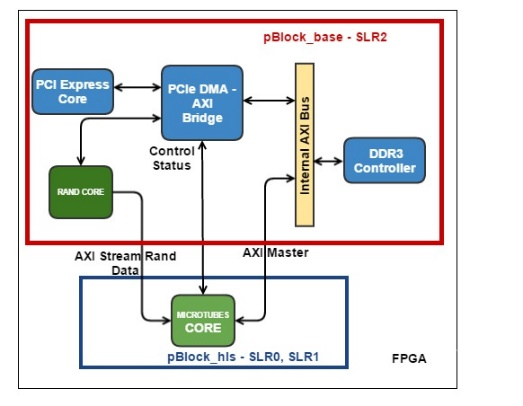
Рис. 3. Блок-схема проекта FPGA. Синим и желтым цветами отмечены блоки, входящие в BSP. Зеленым обозначены вычислительные HLS ядра. Также обозначено разбиение ядер проекта на блоки (pBlocks) для наложения пространсвенных ограничений при трассировке.
Для создания ускоренного приложения была разработана методология, состоящая из нескольких шагов. Во-первых, оригинальный последовательный код компилировался в среде Vivado HLS, и проверялось, что скомпилированный таких образом код не изменяет выходных данных опорного последовательного кода. Во-вторых, из этого кода выделялась основная вычислительная и подходящая для ускорения часть; эта часть отделялась от основного кода с помощью функции-обертки. После чего создавалось две копии такой функции и логика проверки на соответствие результатов обеих частей. Первая копия была опорной реализацией алгоритма в Vivado HLS, а вторая была оптимизирована для трансляции в RTL код. Оптимизации включали в себе переписывание кода, такие как использование статических массивов вместо динамических, использование специальных функций для ввода/вывода в HLS ядро, методы экономии памяти и переиспользования результатов вычислений. После каждого изменения результат функции сравнивался с результатом опорной реализации. Другим методом оптимизации было использование специальных директив Vivado HLS, не меняющих логическое поведение, но влияющих на конечную производительность RTL кода. На данной стадии следует оставаться до тех пор, пока не будут получены удовлетворительные предварительные результаты трансляции C в RTL, такие как производительность схемы и занимаемые ресурсы.
Следующая стадия – это имплементация разработанного вычислительного ядра в системе Vivado вне контекста основного проекта. Здесь задача добиться отсутствия временных ошибок уже разведенного дизайна внутри разработанного вычислительного ядра. Если на этом этапе наблюдаются временные ошибки, то можно применять другие параметры имплементации, либо возвращаться на предыдущую стадию и пытаться изменить С++ код или использовать другие директивы.
На следующей стадии необходимо имплементировать вычислительное ядро уже вместе с основным проектом и его временными и пространственными ограничениями. На данной стадии также необходимо добиться отсутствия временных ошибок. Если они наблюдаются, то можно либо изменить частоту работы вычислительной схемы, наложить другие пространственные ограничения на размещение схемы на кристалле, либо опять заняться
изменением C++ кода и/или использовать другие директивы.
Последняя стадия разработки – это проверка на соответствие результатов, полученных на реальном запуске в железе и с помощью опорной модели на CPU. Проходит она на небольшом промежутке времени, при этом считается, что на более длительных запусках (когда сравнить c CPU уже проблематично) FPGA решение выдает правильные результаты.
Работа в среде Vivado HLS
В работе использовалось два Vivado HLS ядра (Рис. 3): основное ядро, реализующее алгоритм молекулярной динамики микротрубочек (MT ядро), и ядро для генерации псевдослучайных чисел (RAND ядро). Нам пришлось разделить алгоритм на два вычислительных ядра по следующей причине. Кристалл FPGA Virtex-7 2000T – это самый большой кристалл FPGA семейства Virtex-7 на рынке. Он на самом деле состоит из четырех кристаллов кремния, соединенных на подложке множеством соединений и объединенных в один корпус микросхемы. По терминологии Xilinx каждый такой кристалл называется SLR (Super Logic Region). При использовании таких больших FPGA всегда возникают проблемы с цепями, пересекающими границы SLR. Xilinx рекомендует вставлять регистры на такие цепи с обеих сторон границы SLR.
Полное HLS ядро, включающее и MT и RAND ядра, требовали аппаратных ресурсов больше, чем было доступно в одном SLR, поэтому были цепи, которые пересекали границу независимых кристаллов кремния. На стадии трансляции с языка C++ в RTL Vivado HLS ничего «не знает» о том, какие цепи будут впоследствии пересекать границу, и поэтому не может заранее вставить дополнительные регистры синхронизации. Поэтому мы приняли решение разделить ядра на два, пространственно ограничить их в разные SLR и вставить регистры синхронизации на интерфейсные цепи между ядрами на уровне RTL.
Ядро MT
Данный алгоритм очень хорошо подходит для реализации на FPGA, потому для сравнительно небольшого количества данных из памяти (координаты двух молекул) необходимо вычислить сложную функцию сил взаимодействия и удается построить длинный вычислительный конвейер.
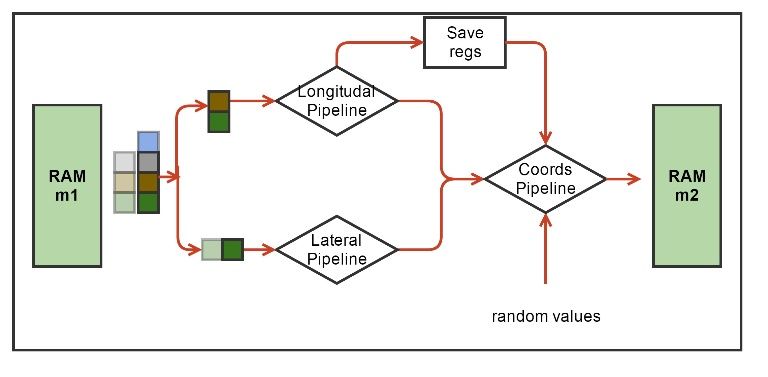
Рис. 4. Блок-схема аппаратной вычислительной процедуры ядра MT. Зеленым обозначены аппаратные блоки памяти для хранения координат молекул. Обозначены вычислительные конвейеры сил и обновления координат, а также блок Save Regs для хранения промежуточных результатов вычислений. Псевдослучайные числа поступают в конвейер обновления координат из другого HLS ядра.
Каждая молекула, т.е. мономер тубулина, взаимодействует только с четырьмя своими соседями (Рис. 2). На каждой итерации по времени надо сначала вычислить силы взаимодействия, а затем обновить координаты молекул. Функции вычисления сил взаимодействия включают в себя множество арифметических, экспоненциальных и тригонометрических операторов. Нашей первой задачей было синтезировать конвейер для этих функций. Рабочим типом данных был вещественный тип float. Vivado HLS синтезировала такие функции в виде конвейеров, работающих на частоте 200 МГц, с латентностью порядка 130 тактов. При этом конвейеры были однотактовые (или, как говорят, с интервалом инициализации равной 1), что означает, что на вход они могли принимать координаты новых молекул каждый такт, а затем после начальной задержки (латентности) – выдавать обновленные значения сил также каждый такт. Выходные силы взаимодействия использовались для обновления координат, что тоже было конвейеризовано. Для обновления каждой координаты каждой молекулы были необходимо независимые псевдослучайные нормально распределенные числа, получаемые из другого HLS ядра. Если взять три молекулы («текущую», «левую» и «верхнюю») то получилось возможным объединить конвейеры вычисления сил и обновления координат в один конвейер, реализующий все вычисления для одной молекулы. Такой конвейер имел латентность равную 191 такт (Рис. 4).
Алгоритм проходит по всем молекулам в цикле. На каждой итерации цикла необходимо иметь координаты трех молекул: одна молекула рассматривается как «текущая», также есть «левая» и «правая» молекулы. Соответственно рассчитываются силы взаимодействия между этими тремя молекулами. Далее при обновлении координат текущей молекулы левая и верхняя компоненты сил взаимодействия брались из расчета на текущей итерации, а нижняя и правая компоненты брались либо из граничных условий, либо с предыдущих итераций из локального регистрового файла Save Regs (Рис. 4).
Количество молекул N в системе было небольшим (13 протофиламентов х 12 молекул = 156 молекул). На каждую молекулу требуется 12 байт. Схема использовала два массива координат m1 и m2, общим объемом меньше 4 КБ, соответственно эти данные легко помещались во внутреннюю память FPGA – BRAM, реализованную внутри HLS ядра. Схема была устроена таким образом, что на четных итерациях по времени координаты считывались из массива m1 (и записывались в m2), а на нечетных – наоборот. С точки зрения алгоритма можно было читать и писать в один массив координат, но Vivado HLS не могла создать схему, способную на одном и том же такте читать и писать один и тот же аппаратный массив, что требуется для работы однотактового конвейера. Поэтому было принято решение удвоить количество независимых блоков памяти.
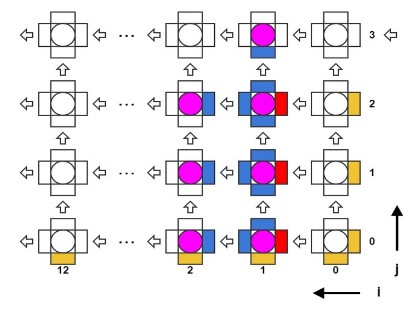
Рис. 5. Схема конвейерного расчета взаимодействий тубулинов в микротрубочке.
Оказалось возможным реализовать три полных параллельных конвейера, способных обновлять координаты трех молекул каждый такт (Рис. 5). Тогда во избежание простаивания конвейеров необходимо было увеличить пропускную способность к локальной памяти и читать координаты семи молекул каждый такт. Это проблема легко решилась, практически не меняя исходный C++ код, а лишь за счет использования специальной директивы,
физически разбивающей исходный массив данных по четырем независимым аппаратным
блокам памяти. Т.к. память BRAM в FPGA является двупортовой, то из четырех блоков памяти можно прочитать 8 значений за такт. Но, так как три конвейера требуют координаты 7ми молекул за такт (см рис 5), это решило проблему.
#pragma HLS DATA_PACK variable=m1, m2
#pragma HLS ARRAY_PARTITION variable=m1, m2 cyclic factor=4 dim=2| Период | L | II | BRAM | DSP | FF | LUT | Утилизация |
|---|---|---|---|---|---|---|---|
| 5 нс | 191 такт | 1 такт | 52 | 498 | 282550 | 331027 | Абсолютная |
| 2 % | 23 % | 11 % | 27 % | Относительная |
Табл. 1: Производительность и утилизация схемы HLS с тремя полными конвейерами
В табл. 1 приводится утилизация схемы HLS (т.е. количество потребляемых ей аппаратных ресурсов FPGA, в абсолютных и относительных единицах для кристалла Virtex-7 2000T) и ее производительность. L – это задержка или латентность схемы, т.е. количество тактов между подачей в конвейер первых входных данных и получением первых выходных данных, II – это интервал инициализации (или пропускная способность) конвейера, означающее через сколько тактов на вход конвейера можно подавать следующие данные.
Утилизация приводится как в абсолютных величинах (сколько требуется триггеров FF или таблиц LUT) для реализации схемы, так и в относительных к полному количеству данного ресурса в кристалле. Как видно из табл. 1 латентность L полного конвейера была равна 191 такту, при этом каждый конвейер должен был обработать третью часть все молекул, что дает теоретическую оценку времени вычисления одной итерации равную T(FPGA) = (L+N/3)*5нс=1.2 мкс
Из табл. 1 также видно, что в кристалле осталось еще много неиспользованной логики, но дальше увеличивать количество параллельных конвейеров непрактично. Будет уменьшаться только второе слагаемое, а начальная задержка все равно будет давать значительный вклад во время работы. При этом увеличение количество логики усложнит размещение и трассировку схемы на следующих стадиях разработки проекта в Vivado.
Ядро RAND
Как было указано, алгоритм учитывает Броуновское движение молекул, одним из методов расчета которого является прибавление нормальной случайной добавки к изменению координат на каждой итерации по времени. Необходимо очень много нормально распределенных случайных чисел, на каждую итерацию – по N3 чисел, что дает поток 420 10^6 чисел/с. Такой поток не может быть загружен с хоста, поэтому его необходимо генерировать внутри FPGA «на лету». Для этого, как и в опорном коде для CPU, был выбран генератор вихрь Мерсенна, дающий равномерно распределенные псевдослучаные числа. Далее к ним применялось преобразование Бокса-Мюллера и на выходе получались нормально распределенные последовательности. Исходный открытый код вихря Мерсенна был модифицирован для получения аппаратного конвейера с интервалом инициализации в 1 такт. Алгоритм требует 9 нормальных чисел каждый такт, поэтому ядро RAND включало в себя 10 независимых генераторов вихря Мерсенна, т.к. преобразование Бокса-Мюллера требует два равномерно распределенных числа для получения 2х нормально распределенных. В табл. 2 приводится утилизация ядра RAND.
| BRAM | DSP | FF | LUT | Утилизация |
|---|---|---|---|---|
| 30 | 41 | 48395 | 64880 | Абсолютная |
| 1.2 % | 9 % | 0.1 % | 5.3 % | Относительная |
Табл. 2: Утилизация ядра RAND
Видно, что такое ядро требует значительную часть DSP ресурсов кристалла, и это ядро было бы сложно разместить в одном SLR с ядром MT, т.к. сумма утилизаций двух ядер хотя бы по DSP ресурсу 31% больше чем может вместить один SLR (25 %).
Более подробно о том, как мы выбирали генератор псеводслучайных чисел и синтезировали его в Vivado HLS можно почитать в статье моей коллеги https://habrahabr.ru/post/266897/
Создание битстрима
После интеграции вычислительных ядер в Vivado на проект были наложены пространственные ограничения на размещение IP блоков. Используемая FPGA Virtex-7 2000T имеет 4 независимых кристалла кремния (SLR0, SLR1, SLR2, SLR3). Было показано, что ядро MT не умещалось в один SLR, поэтому было решено создать два региона размещение (pBlock): pBloch_hls для размещения только MT ядра и pBlock_base для размещения остальных ядер проекта (Рис. 3). Регион размещения pBlock_hls включал в себя SLR0 и SLR1, pBock_base – SLR2. Такой подход позволил разместить логику оптимальным образом, вставить регистры синхронизации на интерфейсы, пересекающие регионы размещения (а значит и SLR) и добиться положительных времянных результатов после трассировки проекта.
Формат хабра позволяет подключать много картинок, поэтому вот еще один рисунок, как в итоге прошла имплементация проекта.
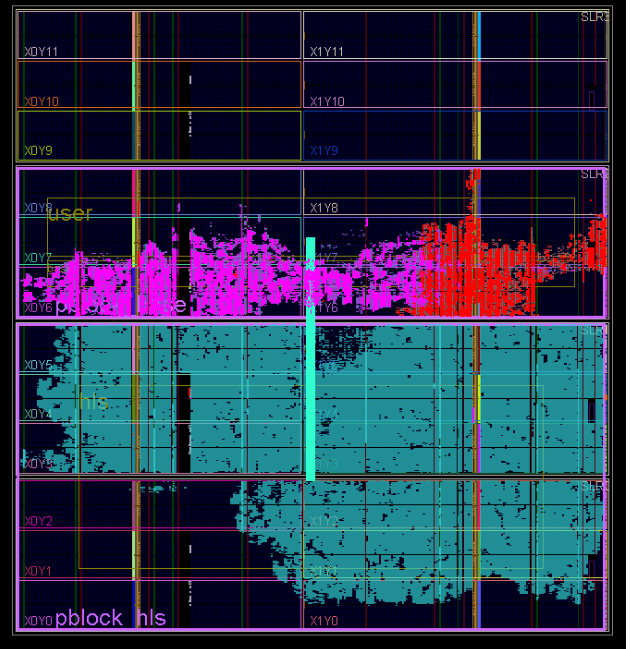
Красным на картинке подсвечены элементы Board Support Package (PCIe core, DDR3 Interface, Internal AXI Bus), голубым — MT HLS ядро, а пурпурным — элементы ядра RAND.
Результаты
Производительность
Результаты работы всех трех реализаций (CPU, GPU и FPGA) были логически верифицированы относительного оригинального кода и признаны состоятельными. Сравнение производительностей производилось замером времени работы программ на 10^7 итераций алгоритма и вычислением времени, требующегося для расчета одной итерации. При этом производительность GPU и FPGA платформ брали в расчет время передачи данных между хост-компьютером и ускорителем.
Для оценки производительности обычно используется метрика операций в секунду. Для данного алгоритма нам оказалось сложным вычислить точное значений вещественных операций, поэтому мы просто сравниваем времена работы алгоритма для вычисления одной итерации, определяя производительность CPU платформы равной 1. Результаты сравнения приводятся в табл. 3, во втором столбце которой приводятся времена вычисления одной итерации алгоритма в микросекундах, а в третьем – относительная производительность платформ.
| Платформа | Время, мкс | Производительность |
|---|---|---|
| CPU | 22 | 1 |
| GPU | 14 | 1.6 |
| FPGA | 1.3 | 17 |
Табл. 3: Сравнение производительности трех платформ
Из таблицы видно, что реализация на GPU быстрее CPU всего в 1.6 раза, в то время как FPGA быстрее CPU в 17 раз. Это означает, что FPGA быстрее GPU в 11 раз. Полученное экспериментально время работы FPGA равно 1.3 мкс на итерацию больше расчетного времени в 1.2 мкс из-за учета накладных расходов на передачу данных по шине PCI Express.
Энергоэффективность
Для измерения энергопотребления мы использовали следующие средства. Для CPU платформы – утилиту Intel Power Gadget. Для GPU платформы — утилиту Nvidia-smi. Для FPGA – специальные программно-аппаратные средства, включенные в состав блока RB-8V7. Во всех случаях замерялась разница в потреблении всего чипа до запуска задачи и во время вычислений. Результаты приведены в таблице 4.
| Платформа | Мощность, Вт | Ex | Ex_rel |
|---|---|---|---|
| CPU | 89.6 | 0.011 | 1 |
| GPU | 67 | 0.023 | 2 |
| FPGA | 9.6 | 1.77 | 160 |
Табл. 4: Сравнение энергопотребления вычислительных платформ
Во втором столбце таблицы приводится мощность, выделяющаяся при расчете на разных платформах. В третьем столбце приводятся значения абсолютной энергоэффективности (производительности на Вт) для данной задачи, определяемой по формуле:
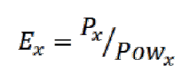
В четвертом столбце приводятся значения относительной энергоэффективности разных платформ для данной задачи, вычисляемой по формуле:
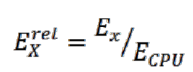
Для обеих формул, x = {CPU, GPU, FPGA}.
Видно, что у FPGA есть большое преимущество в энергоэффективности перед другими платформами, что может сыграть роль в средне и долгосрочной перспективе использования FPGA ускорителей в датацентрах при оплате счетов за электроэнергию. Достигается это в первую очередь за счет того, что FPGA работают на порядок меньшей частоте.
Обсуждение
В ранее опубликованных работах технология FPGA неоднократно применялась к решению задач молекулярной динамики [12]–[17]. Исследователям из лаборатории CAAD Бостонского Университета удалось разработать эффективное ядро для расчета короткодействующих межмолекулярных сил, которое было реализовано на плате ProcStar-III (производство фирмы Gidel), с установленным кристаллом FPGA Altera Stratix-III SE260. Плата имела PCI Express интерфейс к хост-компьютеру. Было показано, что разработанное ускоренное решение было в 26 раз быстрее чистой реализации на CPU на бенчмарке Apoal. В работе [24] авторы перенесли часть пакета для расчета молекулярной динамики LAMMPS на FPGA. Ускоренная часть включала в себя вычисления дальнодействующих взаимодействий. Разработанное аппаратное ядро состояло из четырех одинаковых независимых конвейеров, работающих параллельно. Задача была выполнена на суперкомпьютере Maxwell, каждый узел которого состоит из одного процессора Intel Xeon и двух кристаллов FPGA Xilinx Virtex-4 [25]. Авторы заявили, что разработанное ускоренное решение легко масштабировалось на множество узлов суперкомпьютера Maxwell. Из анализа производительности только ускорителя следовало, что на двух узлах компьютера можно было получить ускорение в 13 раз по сравнению с чисто программным решением. Однако полное время работы гибридного решения было хуже чисто программного из-за того, что время на пересылку данных между CPU и внешней памятью SDRAM, подключенной к FPGA занимало 96% времени работы всего алгоритма. Но в работе утверждается, что если улучшить интерфейс передачи данных, то можно получить полный выигрыш в скорости в 8-9 раз.
В настоящей работе мы применили FPGA к расчету движения ансамбля белковых молекул методом броуновской динамики. Наша программно-аппаратная реализация алгоритма деполимеризации микротрубочки показала, что производительность FPGA при расчете одной траектории микротрубочки в 17 раз превосходила производительность CPU и в 11 раз производительность GPU. Полученное ускорение при расчете деполимеризации микротрубочки методом броуновской динамики позволяет осуществить расчет на временах порядка нескольких десятков и даже сотен секунд. Это позволит предсказывать поведение реальных микротрубочек на экспериментально доступных временах и проанализировать механизмы динамической нестабильности микротрубочек, что будет предметом будущей работы в данном направлении.
Полученный выигрыш на задаче броуновской динамики позволяет говорить о перспективности применения FPGA для решения данного типа задач. Насколько нам известно, это первая попытка сравнить производительность и энергоэффективность различных типов аппаратных ускорителей на данном алгоритме.
Долгое время главной проблемой использования FPGA являлось отсутствие высокоуровневых средств программирования. Традиционные языки описания аппаратуры всегда требуют значительного времени для реализации алгоритма, в то время как первые высокоуровневые трансляторы [26] генерировали RTL код низкого качества. Однако, несколько лет назад компании Altera и Xilinx стали уделять значительные ресурсы этой проблеме и выпустили на рынок свои высокоуровневые средства программирования (Altera SDK for OpenCL и Xilinx Vivado HLS). Данные трансляторы генерируют намного более эффективный код и позволяют прикладному программисту использовать языки C/C++ (Xilinx) и OpenCL (Altera) для создания качественных аппаратных вычислительных схем. В последнее время появилось множество работ, в которых использовались средства высокоуровневого синтеза для разработки FPGA ускорителей [27]–[29]. Например, в работе [28] с помощью средства Vivado HLS реализован алгоритм оптического потока на платформе Xilinx Zynq-7000. Разработанная система имела производительность сравнимую с реализацией на CPU, при этом потребление энергии было в 7 раз меньше. Авторы особенно подчеркивали, что использование средств HLS по сравнению с традиционными RTL языками значительно сократило срок разработки. Использование средства Vivado HLS в ходе выполнения настоящей работы также позволило значительно сократить время и трудоемкость разработки и привлекать к программированию разработчиков, не владеющих специальными навыками работы с FPGA. Все это позволяет говорить об FPGA как о состоявшейся платформе для высокопроизводительных вычислений в области молекулярной и броуновской динамики.
Фуф, спасибо за внимание! Признательности и ссылки на литературу можно найти в оригинальном pdf-документе, доступном на сайте Трудов ИСП РАН
Комментарии (64)
Centimo
21.11.2016 18:29Правильно ли я понимаю, что в итоге вы сравнивали производительность решения на CPU и решения на CPU + FPGA? Или же в последнем случае CPU не производил каких-либо значимых вычислений?

urock
21.11.2016 19:02Мы сравнивали выполнение одинаковых кусков кода — вычисление динамики. Причем делали все честно — с учетом передачи данных между памятью и FPGA.

EqualsZero
21.11.2016 19:03Тема сама по себе интересная, но что именно являлось целью работы?
Если именно исследование микротрубочек, то почему настолько маленькое количество частиц? Может я ошибаюсь, но судя по вики, микротрубочки подлиннее вообще должны быть. Понятно, что как кусочек посмотреть — можно и столько, но может больше — лучше будет?
Если показать использование FPGA для молекулярной динамики, то как я понял, перенести этот подход на другие задачи также эффективно слету не выйдет — используется фиксированое число соседей, ну и опять-таки обычно частиц на порядки больше.
urock
21.11.2016 19:08+1Цели работы были две.
1. Получить реальное ускорение на реальной задаче. Маленькое число частиц — особенность задачи, потому что вычисляются изменения только на конце микротрубочки, а она еще растет или уменьшается (не учитывается в этой реализации, но вы можете посмотреть у нас на гитхабе, мы это допилили), так в любом случае нижняя часть стабильна и не интересна. Сейчас коллеги реально запускают вычисления и скоро опубликуют результаты.
2. Показать, что FPGA применимо в этой сфере, и не только в статьях зарубежных, но и у нас в России. Эта задача — первая ласточка. Скоро будут результаты по использованию метода Монте — Карло как для этой задачи, так и для других, например ряда финансовых задачь.
RomanArzumanyan
21.11.2016 19:05В работе показано, что реализация на FPGA быстрее CPU в 17 раз и быстрее GPU в 11 раз.
Тревожный звонок. Если топовая Tesla в таких задачах не опережает CPU как следует, то что-то пошло не так. Ну и выбор OpenCL для написания кода под Nvidia — это странно.urock
21.11.2016 19:11+2Мы честно говорим, что производительность GPU мала на этой задаче в первую очередь из-за того, что размер задачи мал, и в итоге загрузка GPU очень мала. Что поделать, такая задача. Мы и ищем в первую очередь такие, на которых можно конкурировать с GPU. OpenCL для GPU — это стандарт, который поддерживает Nvidia. Мы проверя на CUDA, было точно также. Дело в том, что OpenCL теперь работает и на FPGA.
RomanArzumanyan
21.11.2016 19:40+1А вы проводили профилировку кода на CPU/GPU? Есть ли аналогичные готовые решения (для CPU должны быть, по идее), которые показывают схожую с вашей производительность?
Иными словами: хотелось бы знать, что за 17-кратным превосходством стоит хорошее решение задачи на FPGA, а не меделенный код для CPU, который можно значительно ускорить. Я не занимался никогда разработкой под FPGA, но (по слухам :) ) это дело небыстрое — что будет, если потратить такое же время на написание оптимизированного кода с использованием intrinsic'ов и ассемблера?urock
21.11.2016 19:45Роман, все что мы делали, я описал. Мы максимально старались сделать параллельный производительный код на CPU. Мы публикуем эти результаты открыто. Очень будем рады, если кто-то возьмется и попробует сделать быстрее как на CPU так и на GPU.
Но про ассемблер ничего не могу ответить, кажется что это сложно. Время разработки FPGA схемы я тут приводил в комментариях — 1.5 месяца отладка вычислительной схемы full time и два месяца расслабленной работы по укладываюнию во времянные ограничения.

mezastel
21.11.2016 20:51Это обычно показывает, что задача плохо ложится на классический SIMT парралелизм и содержит очень много dataflow обработки. Именно на этих сценариях, а также в ситуициях где много бренчинга, по идее FPGA и должен выигрывать.
urock
21.11.2016 21:14Согласен с первым предложением. Но не со вторым =) Под каждую бранч в FPGA создается своя логика, если бранч не выбирается, логика все равно остается, в итоге код с большим количеством веток н будет занимать очень много места, а аппаратура будет простаивать, поэтому выигрыша можно не получить.
Gryphon88
21.11.2016 22:58Расскажите, пожалуйста, побольше про модель, честно говоря, лениво читать [20-21]. Рассматривалась внеклеточная среда и голая физхимия, или клеточная, с учётом кэпирующих белков и прочей нечисти?
urock
22.11.2016 10:25Насколько я, программист понял, модель такая. Есть структура молекул микротрубки, она шевелиться под действием сил взаимодействия между этими молекулами, а также под действием Броуноского движения. Это динамика. Каждый шаг динамики — это 0.2 нс. Ее мы и считали на FPGA. Еще есть кинетика, это вероятностьный процесс присоединения/отсоединения одного димера от трубочки. происходящий на временах порядка 1 мс (могу ошибиться ± один порядок). По сравнению с динамикой, кинетика — это процесс редкий, и поэтому считается на CPU.
Т.е. была среда, где шевелилась микротрубка, которая могла из окружающей среды присоединять свои молекулы, или те могли отрываться. Других белков вокруг не было. Вероятности присоединения/отсоединения задавлись параметрами, физическую суть которых я до конца тут не смогу изложить.Gryphon88
22.11.2016 11:52удалось ли подобрать условия, в которых наблюдается:
— динамическая нестабильность (присоединение и уход мономеров на обоих концах)
— тредмиллинг (рост на одном конце с одновременным укорочением на другом)
— катастрофа с раскрытием МТ как цветка
Все три поведения описаны, но экспериментальное подтверждение условий, когда доминирует тот или иной тип роста, вроде пока не появилось. Уж очень трудоёмко и на грани разрешения аппаратуры.
Ещё такой вопрос: изучалось поведение одиночных МТ, или ещё дуплетов\триплетов?urock
22.11.2016 15:01— Моделировался только один конец
— с катастрофы с раскрытием мы начинали моделирование чтобы верифицировать наш генератор псевдослучайных чисел. Работали только с одной МТ. Вообще говоря, вопросы не ко мне, я могу вас в личке свести с учеными биофизиками, кто ставил задачу и вообще решают ее. Я только FPGA ускоритель =)Gryphon88
22.11.2016 15:111. Поймать Фазли Иноятовича — это неочевидная задача, в точности по Гейзенбергу: или время, или место. Если и то, и другое — то надо бегать по 4 корпусам весь рабочий день. А других биофизиков понять лично мне значительно сложнее, большинство знает или что делали, или зачем. Мне проще Вас поспрошать, Вы-то уж точно с обеими группами общались.:) Ну и статьи я точно прочту, по этой и в сборнике ИСП РАН модель и конечная биологическая цель работы были непонятны.
2.с катастрофы с раскрытием мы начинали моделирование чтобы верифицировать наш генератор псевдослучайных чисел
Эт как? Событие же очень редкое.urock
23.11.2016 10:252. Ну не знаю, просто если задать начальные условия с уже сильно раскрытым венчиком, то трубочка раскрывается очень быстро. Посмотрите видео в этой статье, где моя коллега подробно описала, как мы реализоывали генератор псевдослучайный чисел.
Хотя может я и путаюсь, это не наверное не катастрофа все-таки, а просто динамическая релаксация…
Коллеги из группы Фазли сейчас как раз работают над продолжение моделирования на FPGA, я обязательно упомяну тут об их прогрессе.

kosmos89
22.11.2016 02:16Что-то ваша программа (из папки cpu) вылетает в VS2015 с ошибкой:
Run-Time Check Failure #2 — Stack around the variable 'long_d_t' was corrupted.
OpenMP в компиляторе не включал, если что.kosmos89
22.11.2016 02:27+1Ой, да у вас и вправду программа некорректная!
calc_grad_c принимает последним аргументом &long_d_t[i][j + 1], а на последней итерации этот ваш j+1 указывает за пределы массива!
Почините сначала код свой, а потом производительность измеряйте. Готов спорить, если прямыми руками с пониманием написать, то все ускорится в разы, особенно GPGPU версия.urock
22.11.2016 10:19Компилировали под Линкс с помощью gcc. За указание на ошибку спасибо, посмотрим
ivananashkin
22.11.2016 14:26Насколько FPGA будет опережать GPU в классической молекулярной динамике? Стоит ли ждать отдельных устройств узкой направленности? Например прибор для аппаратного ускорения того же громакса. Насколько я понимаю под каждую отдельную задачу надо заново компилировать программу и зашивать в нее параметры потенциала межмолекулярного взаимодействия и прочие условия моделирования. Так ли это?
DaylightIsBurning
22.11.2016 15:53Эта задача решена институтом D.E. Shaw, они даже дальше пошли — кластер ASICов запилили (Anton, Anton II). Ускорение примерно на два порядка для первой версии ASIC.
DaylightIsBurning
22.11.2016 17:32+2Попробую составить более полную картину. Для молекулярной динамики у меня получается:
производительность одной FPGA=19.4 x 2 x Intel Xeon E5-2640 (2x6 cores), 92.2k atoms
производительность одной GeForce GTX 1080 = 11.4 x 2 x Intel Xeon E5-2650V3 (2x10 cores), 90.9k atoms
Кластер из 512 нод ASICов (Anton II) = 377 x GTX 1080, 23.6k atoms
Кластер из 512 нод ASICов (Anton II) = 941 x GTX 1080, 90.9k atoms
То есть производительность одной GTX 1080 примерно равна производительности одной FPGA. Кластер 512 ASICов производительней на 3 порядка. То есть один ASIC примерно равен по производительности одному GPU, но ASIC масштабируются «в лоб» намного лучше чем GPU. То есть можно взять 512 ASIC и получить производительность в 512 раз выше (в рамках одной симуляции), больше 512, вроде не получится уже. Производительность GPU масштабируется «в лоб» максимум раза в 2-4 ещё на данный момент.
Единственный смысл в Anton, на сколько я могу судить, — это неприрывные длинные симуляции порядка миллисекунд и более, которые на GPU не достижимы. Во многих случаях необходимости в этом нет, так как аналогичные задачи можно решить не «в лоб», а с помощью задействования кучи GPU и enhanced sampling алгоритмов типа REMD или Markov State Models, которые не требуют интенсивной коммуникации между GPU. Три порядка всё равно трудно догнать, но один-два порядка — вполне. Задействовать такие алгоритмы на Anton не выйдет — дорого по деньгам.
По цене кластер ASICов совершенно заоблачный (малосерийный). В открытую Anton не продают, но на конференциях ходили слухи что они готовы продать такой кластер за примерно $10 млн. Одна GTX1080 стоит 600$, в ноде будет порядка 700-800$ с учётом материнки, ЦПУ и т.п.
В плане гибкости, GPU, конечно, намного предпочтительней, как и в плане обслуживания. Софта для MD на GPU полно, в т.ч. бесплатного. Специалистов, знакомых с GPU, тоже полно (чего не скажешь про custom-made ASIC clsuter). Соответсвенно доступность разных «расширений» за пределами базовой стандарной МД на GPU намного лучше.
CPU/GPU
FPGA/CPU
ASIC cluster (Anton II)ivananashkin
22.11.2016 18:01Спасибо за ответ. Мы исследуем мембранные процессы разделения в неравновесных условиях молекулярной динамикой и указанные алгоритмы нам не подходят. Мы как раз проводим длительное моделирование, один только выход на стационарность может занимать несколько месяцев реального времени на нескольких тысячах молекул.
Ну и как не прискорбно это признавать, в нашем университете компьютеры не считаются средствами научных исследований, и никаких ГТХ1080 на соответствующую статью расхода купить не получится ((. По факту приходилось часть ГПУ покупать за свои. Ладно хоть за немцев порадуюсьDaylightIsBurning
22.11.2016 18:30А почему REMD не подходит для неравновесной МД?
ivananashkin
22.11.2016 21:34Специфика расчетов такая. Мы используем немного модифицированный метод DCV GCMD, и я не знаю как к нему прикрутить REMD.
Psychopompe
22.11.2016 19:20Да за 10 миллионов можно отдельный суперкомпьютер собрать. А я уж было понадеялся, что FPGA это дешёвый и сердитый способ работать с MD.
DaylightIsBurning
22.11.2016 19:35дешёвый и сердитый способ работать с MD — это GPU. С несколькими GPU уже можно набрать материал достаточный для публикации в нормальных научных журналах.
D.E.Shaw research по сути спроэктировали и собрали special-purpose суперкомпьютер и специализорованный же процессор, отсюда и цена. D.E. Shaw вообще заработал деньги (миллирды, похоже) на разработке специализированных суперкомпьютеров для финансов/биржи в 90е. Затем ушел из финансов и на заработанные деньги финансирует собственный исследовательский институт, занимается вычислительной биохимией в основном.
D.E. Shaw вообще заработал деньги (миллирды, похоже) на разработке специализированных суперкомпьютеров для финансов/биржи в 90е. Затем ушел из финансов и на заработанные деньги финансирует собственный исследовательский институт, занимается вычислительной биохимией в основном.
Ожидать что FPGA будет дешевле/быстрей GPU было излишне оптимистично — GPU в MD загружаются на полную, но при этом они массовые в производстве за счёт геймеров. Это подтверждается также тем, что кастомный ASIC разработанный специально для MD по производительности равен GPU.
anprs
23.11.2016 10:13А если какой-нибудь NVIDIA Jetson TX1? Или кластер собрать из 128 Raspberry PI Zero?
DaylightIsBurning
23.11.2016 12:02На счёт Jetson точно не скажу, но raspberry не пойдет — слишком медленно. Что бы догнать один GPU, приходится 64 ядра Ксеонов задействовать, 128 малин такой производительности и близко не дадут, не говоря уж о совершенно недостаточной скорости интерконекта, в кластерах ЦПУ всякие супер производительные InfiniBand применяют, и то еле-еле хватает.
Psychopompe
24.11.2016 04:54Я думал о GPU коде для своей задачи, но у меня не полноценная MD, так что я пока не уверен, что лучше использовать.
DaylightIsBurning
22.11.2016 17:44Создавать прибор для аппаратного ускорения MD на базе отдельного FPGA смысла, на мой взгляд, нет т.к. GPU не медленнее, удобнее, проще и уже всё готово и оптимизировано.
old_bear
23.11.2016 00:15+1Безусловно нет смысла в попытках заменить GPU на FPGA путём эмуляции этого GPU на FPGA.
Но если есть возможность использовать гибкость FPGA, то всё может быть более интересным. Например, может быть интересной идея создать внутри FPGA объектное представление моделируемой системы, с нужными свойствами объектов и буквальными связями. Типа нейросети, хотя это может быть не самым удачным примером, т.к. нейросети из простых элементов уже научились моделировать на GPU весьма успешно.
urock
23.11.2016 10:19Главное преимущество FPGA — это возможность собрать длинный вычислительный конвейер, каждая ступень которого будет рабоать каждый такт над новыми данными, а сами данные последовательно проходить каждый такт через конвейер. Как было сделано в нашей реализации.
DaylightIsBurning
23.11.2016 13:13В случае МД всё упирается в количество FLOPS в конечном итоге, а оно зависит от транзисторного бюджета и частоты. По совокупности этих параметров GPU сильно опережает FPGA. Польза FPGA в том, что можно на полную загрузить все транзисторы там, где GPU полностью загрузить не получается. Но МД позволяет эффективно загружать GPU, так что я не вижу перспектив у FPGA, что подтверждается и соотв. попытками. Я уже давал ссылку, где люди закодили MD на 4 x Xilinx V5 и догнали один GPU. То же самое подтверждается тем, что даже команда довольно крутых спецов на многомиллионном бюджете смогла лишь догнать по производительности GPU, запилив специальный ASIC. Преимущество ASIC в том, что там можно более эффективный интерконнект организовать и улучшить масштабируемость, но не удельную производительность.

ndv79
23.11.2016 10:15Тут что-то не так: на вашей GPU должно выполняться на порядок быстрее, чем на вашем CPU. Возможно, у вас расходятся потоки, или слишком много используется переменных и как результат они выделяются в глобальной памяти, или вы не используете память local, или вы недостаточно хорошо распараллелили свой алгоритм, или слишком часто используете барьер, или еще что-то.
Да, на всякий случай: OpenCL — хороший выбор для программирования GPU, он не медленнее CUDЫ.urock
23.11.2016 10:17Я думаю, что GPU плохо справляется из-за узкого горла к общей памяти, в которой находят массивы генератора псевдослучайный чисел.
ndv79
23.11.2016 11:26Рандомные числа можно сгенерировать с помощью get_global_id() и побитовых операций. См пример тут: http://stackoverflow.com/questions/9912143/how-to-get-a-random-number-in-opencl
urock
23.11.2016 14:19Я боюсь, что период таких генераторов будет недостаточным для задачи моделирования Броуновского движения. Мы реализовывали известный генератор "вихрь Мерсенна", имеющий период около 2^19937
old_bear
24.11.2016 15:59В отдельной статье по генератору, на которую вы давали ссылку в тексте, в комментариях посоветовали весьма простой и эффективный PCG. Он вам по каким-то причинам не подошёл или просто не захотели менять собственноручно накоденное, пусть и в ущерб производительности?
urock
24.11.2016 16:03Нас уже устраивало то, что было реализовано. У нас не было задачи перебрать множество генераторов и выбрать лучший для задачи, достаточно было найти первый работающий.
old_bear
24.11.2016 16:07Чуть выше вы сами предположили, что ваш вариант генератора является узким местом в реализации на GPU из-за доступа к памяти. Т.е. вас устраивает, что GPU-реализация вероятно работает сильно медленнее чем это возможно?
Если честно, то в этом вашем ответе (и некоторых других ответах) проскальзывает нежелание потратить некоторое количество времени на оптимизацию вашего же (не обязательно лично вашего, но вашей команды) кода. При том что речь может идти не о процентах, а о разах ускорения. Выглядит немного странно.urock
24.11.2016 16:15-1Я вас не пойму. Генератор вирхь мерсенна изначально использовался в коде, который был у ученых. Мы попробовали сделать свой на основе центральной предельной теоремы — не вышло, был плохой период, трубочка разваливалась. В итоге запилили вихрь мерсенна, с ним все было ок. Эту же версию и портировали на GPU. Вы меня обвиняете в том, что я потратил мало времени на вылизование кода GPU? И даже не исследовал, как поведут себя другие генераторы на GPU.
Еще раз повторяю, у меня не было такой задачи.
Что плохого в том, что я хочу показать выгодную мне технологию в хорошем свете? Я же не говорю, что GPU всегда хуже, я говорю на данном алгоритме, который придумал не я, FPGA показывают себе лучше, вот и все.old_bear
24.11.2016 16:25Ок, возможно проблемы не у вас, а у того, кто ставил задачи перед вами и другими разработчиками.
DaylightIsBurning
24.11.2016 18:32Вы пишите:
Это означает, что FPGA быстрее GPU в 11 раз.
Это некорректно. Быть может, было бы корректней сказать, что наивный порт на GPU в 11 раз медленнее вашей реализации на FPGA? Вы для демонстрации выбрали алгоритм генерации псевдослучайных чисел, который был разработан под CPU. Существуют алгоритмы (и реализации) для генерации псевдослучайных чисел для GPU, которые ничем не уступают Мерсенну и хорошо работают.old_bear
24.11.2016 22:34Ну в общем то автор достаточно прямым текстом обозначил свою мотивацию в этом сообщении.
kosmos89
23.11.2016 13:10Скажите, а в чем вообще проблема подождать четыре секунды? Зачем вы мучаетесь с OpenMP, OpenCL, FPGA?
Наверное вам надо посчитать больше, чем одна трубочка? Раскройте, пожалуйста, а люди, возможно, подскажут как это лучше распараллелить.urock
23.11.2016 14:20+1Подождать 4 секунды проблемы нет. Но ученым надо моделировать порядка 200 сек времени жизни МТ, а это с шагом 0.2 нс раньше выливалось в месяцы непрерывного счета. Теперь это дни.
old_bear
(голос с галёрки fpga-дизайнеров)
интересно, как быстро это будет работать, если написать код на hdl-е достаточно прямыми руками…
urock
Честно говоря, я не уверен, что это можно вообще написать на rtl за приемлемое время (скажем полгода). Говорю как RTL программист со стажем. Что до сравнения HLS и ручного кодирования, то уже давно вышла куча статей, где эти подходы сравниваются, и утрверждается, что HLS даже лучше делает. Потому что позволяет вам из одного C кода с помощью прагм получать разные архитектуры, в итоге вы можете выбрать в зависимости от ваших задач.
Один из примеров
old_bear
За полгода на RTL можно написать практически всё. Это я вам говорю как FPGA-дизайнер (не программист) со стажем.
Не знаю, что там за «куча статей», но за последние 15 лет я ни разу не видел, чтобы HLS давал лучшие (или сопоставимые) результаты, чем рационально написанный RTL, если речь не идёт про «синтез» путём подстановки готовых, вусмерть оптимизированных в RTL-е, библиотек.
Что HLS даёт результаты быстрее — видел. Что HLS даёт в несколько раз больший размер в кристалле — видел. Что HLS даёт частоту на 30-100% ниже — тоже видел. А вот чтобы HLS давал сопоставимые с RTL-ем результаты — увы. Но обратите внимание, я имею ввиду RTL, который как RTL, а не как С с подправленным под verilog синтаксисом.
Но я не буду спорить, что для быстрого получения результатов в ограниченном наборе областей HLS уже более-менее годен. Кстати, сколько приблизительно человеко-часов вы потратили на FPGA-часть этого проекта?
urock
Ок, я не готов обсуждать производительность RTL разработчиков, в любом случае есть гении, а есть посредственные.
Надо просто четко понимать область применимости HLS. Это дизайны с потоковыми вычислениями, длинными вычислительными конвейерами, дизайны где не критична latency, где регулярный доступ к памяти.
Основной код схемы я писал один примерно месяц-полтора в full time, это вместе с логическим верифицированием. Дальше были проблемы с таймингами, уже не связанные с HLS, а со структурой микросхемы, состоящей из 4 отдельных кристаллов, которые мы решали еще пару месяцев но уже в расслабленном режиме, переписываясь с инженерами Xilinx.
UPD. Да и приличный и доступный HLS транслятор Vivado HLS появился в 2013, а не 15 лет назад.
old_bear
Вопрос не в гениальности, а в подходе.
Если пишется RTL, то подразумевается, что вы сами выбираете архитектуру, которая реализует алгоритм, и имеете над ней полный контроль. И отличие RTL-дизайнера от программиста как раз в том, что он имеет опыт выбора правильной архитектуры, которая обеспечит нужные характеристики по скорости и размеру. Он может уметь это лучше или хуже, но он это умеет.
В случае же HLS-а вы этого лишены полностью или большей частью. И может наступить такой момент, когда вроде всё должно работать, но «не лезет» по тому или иному критерию. И очень хорошо, если есть возможность эту проблему обойти и весь проект не выкидывается в корзину со словами вида «скорость вычислений не удовлетворяет условиям заказчика».
Так что быстрое прототипирование при условии большого запаса по ресурсам — наверное, да. А что-то зажатое более строгими требованиями — пожалуй, нет.
nerudo
Не совсем так. Высокоуровневые средства все же весьма продвинулись за последнее время. Имеется большое количество настроек реализации. В RTL вы становитесь заложником выбранной «архитектуры» и если просчитались — вынуждены руками переписывать все с нуля. HLS же дает возможность перебирать те самые архитектурные решения, выбрав подходящее вам.
old_bear
Слово «архитектура» вы заметили в моём комментарии. Но заметили ли вы слова «полный контроль»?
Выбрать правильную архитектуру, опираясь на достаточно конкретные расчёты и опыт — это и есть суть работы RTL-дизайнера. С тем же успехом можно назвать программиста заложником своего кода.
Не думайте, что я не знаком с возможностями HLS-а. Его возможности по выбору «архитектурных решений» весьма узки.
Вот, например, в обсуждаемой статье говорится что «Vivado HLS не могла создать схему, способную на одном и том же такте читать и писать один и тот же аппаратный массив». Когда я писал hdl-код, где была необходимость каждый такт читать-изменять-записывать значение некоего контекста хранящегося в BRAM-е по полностью произвольному адресу, я потратил на это от силы день. Применённое решение работало без потерь по скорости и было достаточно элементарным, но не таким, которое способен выбрать HLS.
Хорошо, когда есть обходные пути для решения различных ограничений HLSа. Но иногда этих путей нет или они приводят к весьма заметным потерям ресурсов или частоты.
urock
Да согласен, с записью и чтением на одном такте HLS показала себя не с лучше стороны. Еще у нас были большие проблемы с однотактовой реализацией «вихря Мерсенна», и только когда я нарисовал схему, я понял как написать С код так, чтобы он выдавал значения каждый такт.
С другой стороны мы использовали версию 2014.4, сейчас в 2016.x версии могло что-то улучшиться.
AleCher
Чуть быстрее, но на железе за существенно меньше денег. Что при трудозатратах на разработку может и дороже получиться.
old_bear
В некоторые моменты между HLS-ом и RTL-ем как раз проходит граница помещается/не помещается в самый большой доступный кристалл. Или, например, в клоковый регион, как это произошло у автора статьи.
urock
Поправка: не в клоковый регион, а в отдельный кристалл SLR. С пересечением клоковых регионов проблем не было.
Halt
Кстати а как это делается на практике? Там на все 4 кристалла объединенные клоковые сети от одного PLL (в пределах домена)? Или буферная синхронизация клоков?
urock
Не совсем, не проблема использовать один и тот же клок в разных SLR, это никто не запрещает. Проблемы с цепями, которые пересекают границу SLR. На них очень большая задержка дополнительная получается. Их очень желательно пайплайнить с обеих сторон границы. На практике это делается так. Вы делаете разные модули и констрейните их размещение в разные SLR, и уже ручками в этих модулях ставляете регистры на интерфейсные сигналы. Так я и сделал, разделил код на два HLS ядра, законстрейнил в разные области и вставил регистры на уровне RTL. Но осталась проблема с основным ядром, которое ну никак в один SLR не влезало. Тут пришлось экспериментировать с разными прагмами во время синтеза в HLS.
old_bear
Чудес не бывает. Если есть задержки на обычных цепях, то точно так же они есть и на клоковых. Подозреваю, что если клок вылазит в соседний SLR, то ощутимо ухудшается тайминг, т.к. софт вынужден рассчитывать на больший разброс работы клокового дерева.